
More Information
Submitted: June 29, 2026 | Accepted: July 07, 2026 | Published: July 09, 2026
Citation: Benmakhlouf R, Chouhani BA, Kouiri O, Allata Y, El Bardai G, Kabbali N, et al. Semantic Cardio Reports Generation Using Generative AI. J Clini Nephrol. 2026; 10(7): 70-81. Available from:
https://dx.doi.org/10.29328/journal.jcn.1001178
DOI: 10.29328/journal.jcn.1001178
Copyright license: © 2026 Benmakhlouf R, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Tuberous sclerosis complex; Second hit; Genotype-phenotype correlation; Prognosis, Case report
Abbreviations: TSC: Tuberous Sclerosis Complex; AML: Angiomyolipoma; mTOR: mechanistic Target Of Rapamycin; CKD: Chronic Kidney Disease; ESRD: End-Stage Renal Disease; LAM: Lymphangioleiomyomatosis; HD: Haemodialysis; GTP: Guanosine Triphosphate; RHEB: Ras Homolog Enriched in Brain; PKD1: Polycystic Kidney Disease 1; HBV: Hepatitis B Virus; HCV: Hepatitis C Virus; HIV: Human Immunodeficiency Virus; ANA: Antinuclear Antibodies; ANCA: Anti-Neutrophil Cytoplasmic Antibodies; NBCA: N-Butyl Cyanoacrylate; eGFR: estimated Glomerular Filtration Rate; BPH: Benign Prostatic Hyperplasia; CT: Computed Tomography; MRI: Magnetic Resonance Imaging

Tuberous Sclerosis Complex with Renal Involvement: Not Always a Family Matter-A Case Report
Rime Benmakhlouf1,2*, BA Chouhani1,2, O Kouiri1,2, Y Allata1,2, G El Bardai1,2, N Kabbali1,2 and T Sqalli Houssaini1,2
1Department of Nephrology-Hemodialysis and Renal Transplantation, Hassan II University Hospital, Fès, Morocco
2Faculty of Medicine, Pharmacy and Dental Medicine of Fez, Sidi Mohamed Ben Abdellah University, Morocco
*Corresponding author: Rime Benmakhlouf, Department of Nephrology-Hemodialysis and Renal Transplantation, Hassan II University Hospital, Fès, Morocco, Email: [email protected]
Background: Tuberous Sclerosis Complex (TSC) is a rare autosomal dominant neurocutaneous disease caused by mutations in the TSC1 or TSC2 tumour suppressor genes. It is characterised by hamartomatous lesions in multiple organs and displays a high degree of phenotypic variability. Renal involvement — present in 50 to 80% of patients — is a major cause of morbidity and mortality, predominantly manifesting as angiomyolipomas (AML) and renal cysts.
Case presentations: We report three unrelated patients with TSC presenting with diverse modes of revelation. Case 1: a 65-year-old man admitted for macroscopic haematuria following abdominal trauma, with incidental discovery of severe chronic renal failure, bilateral renal AMLs complicated by a right parapyelic haematoma, cardiac rhabdomyoma, retinal astrocytoma, and calcified subependymal nodules. Case 2: a 45-year-old man with a long-standing TSC diagnosis presenting for management of facial angiofibromas and stable chronic renal failure on a single kidney following prior nephrectomy. Case 3: a 14-year-old boy admitted for hypertensive emergency and advanced bilateral cystic renal failure with rapid progression to end-stage renal disease.
Diagnoses, interventions & outcomes: All three patients fulfilled the 2021 international TSC diagnostic criteria on clinical and radiological grounds, without genetic confirmation. Management included selective arterial embolisation in Case 1, ablative CO2 laser therapy and topical Sirolimus in Case 2, and emergency haemodialysis with corticosteroid therapy in Case 3. Cases 1 and 3 progressed to end-stage renal disease; Case 2 maintained stable renal function over a ten-year follow-up.
Conclusion: This case series highlights the phenotypic heterogeneity of TSC and the diversity of its renal presentations, reinforcing the importance of systematic multiorgan screening, familial investigation, and tailored multidisciplinary management.
Tuberous Sclerosis Complex (TSC) is a rare héréditaire multisystem disorder with an estimated prevalence of 1 in 20,000 to 25,000 live births worldwide [1,2]. It results from loss-of-function mutations in either the TSC1 or TSC2 genes, leading to constitutive activation of the mTOR signalling pathway and the formation of hamartomatous lesions in multiple organs [1]. While the disease is inherited in an autosomal dominant fashion in approximately one third of cases, sporadic de novo mutations account for the majority of presentations [5].
Renal involvement, present in 50 to 80% of patients, is a leading cause of TSC-related morbidity and mortality, primarily through angiomyolipomas, renal cysts, and — more rarely — renal carcinoma [14]. Clinical expression remains highly variable, a phenomenon explained in part by the “Second Hit” somatic mutation theory [28]. This variability poses significant diagnostic and therapeutic challenges.
We herein report three unrelated patients with TSC, each presenting with a distinct mode of revelation and a broad spectrum of renal involvement. This case series aims to illustrate the diversity of clinical presentations, analyse genotype–phenotype correlations, and discuss current therapeutic strategies, while underscoring the value of systematic familial screening.
Patient AS, 65 years old, married and father of 11 children, was admitted to our nephrology department for investigation of an inaugural macroscopic hematuria, non-clotting, of acute course, occurring in the context of a road traffic accident with abdominal trauma.
This hematuria was not associated with irritative or obstructive urinary signs, nor with lumbar pain or renal colic.
No similar previous episode was reported (Table 1a and 1b).
| Table 1a: Case 1 – Summary against the 2021 international TSC diagnostic criteria. | |
| Major criteria present | Minor criteria present |
| Facial angiofibromas; cephalic fibrous plaque; ungual fibromas (≥2); shagreen patch; retinal hamartoma (astrocytoma); subependymal nodules (≥2); cardiac rhabdomyoma; renal AML (≥2) | Multiple renal cysts; non-renal hamartoma (hepatic AML) |
| ≥2 major criteria → clinically definite TSC (genetically unconfirmed). |
| Table 1b: Case 1 – Clinical timeline. | |
| Time | Event |
| Before admission | 15 pack-years tobacco (ceased >20 y); BPH on alpha-blocker; bilateral inguinal hernia repair; no prior TSC diagnosis |
| Admission (age 65) | Macroscopic haematuria after abdominal trauma; bilateral renal AML with right parapyelic haematoma (80×81 mm) and inferior-polar-artery aneurysm; largest cyst 8.1 cm (right lower pole); severe CKD (creatinine 654 µmol/L) |
| Extension work-up | Cardiac rhabdomyoma; retinal astrocytoma; calcified subependymal nodules; hepatic angiomyolipoma |
| Acute treatment | Selective arterial embolisation (NBCA glue); day-3 CT: haematoma stabilised (82×77 mm) |
| Follow-up | Progression to end-stage renal disease → chronic haemodialysis |
| Familial screening | TSC skin lesions in 4 sons + 2 daughters; bilateral renal cysts in 1 son + 2 daughters |
| Genetics | Not performed |
Past medical history
The patient reports chronic smoking for 15 years, having quit more than 20 years ago. He has benign prostatic hyperplasia (BPH) treated with an alpha-blocker. He underwent bilateral surgical repair of an inguinal hernia (Figure 1).
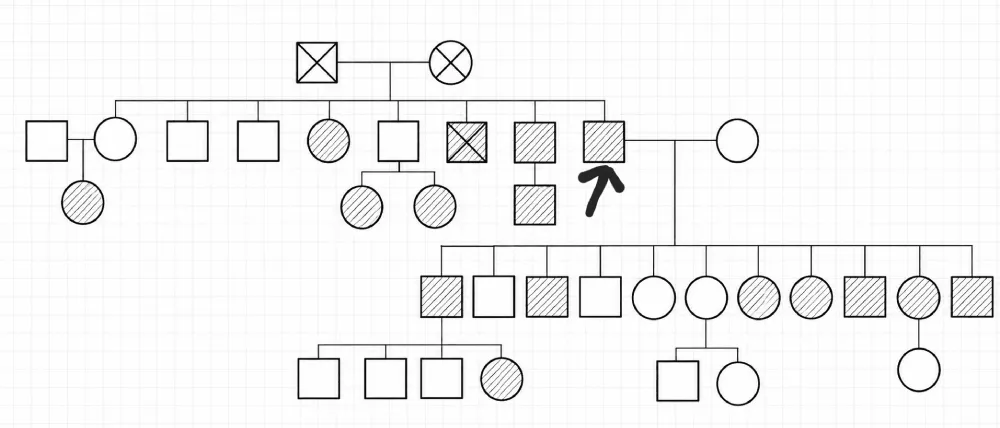
Figure 1: Family tree representing the screening in the family of case 1. Legend : Arrow: designates the patient Square: Male; Circle: Female; Crossed-out symbol: Deceased person; Empty symbol: Healthy person; Hatched symbol: Affected person Horizontal line between two persons: Marriage / Union ┊┊┊ Vertical descending line: Descendants (children)
Clinical examination
On admission, the patient was apyretic, hemodynamically stable and clinically euvolemic. Cardiopulmonary examination was unremarkable. However, abdominal distension was noted without associated transit disorder.
Cutaneous examination revealed pinkish nodular lesions on the mid-face, particularly on the nasolabial folds and cheeks, as well as a few fibrous plaques of approximately 1 cm located on the left frontal area. Diffuse ungual fibromas were observed on the fingernails and toenails. Shagreen patches were also found on the arms and back (Figure 2).
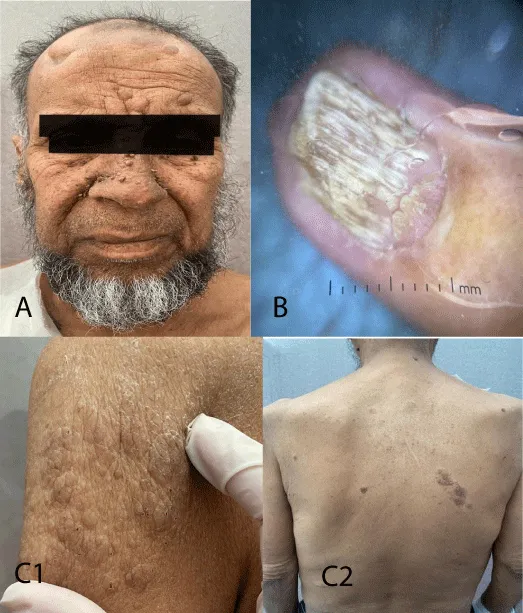
Figure 2: Images of the different dermatological manifestations of the patient (case 1). A: Cutaneous angiofibromas located on the nasolabial folds, cheeks and left frontal area. B: Diffuse ungual fibromas on the fingernails and toenails. C: Shagreen patches on the arms C1 and back C2. Café-au-Lait spots were also found. All of these lesions, corresponding to multiple facial angiofibromas of severe form, were suggestive of a systemic disease. No sensory or motor neurological deficit nor visual acuity disturbance was found.
Labora to work-up
The initial work-up, performed as part of the assessment of overall impact, revealed a urinary cytobacteriological examination with hematuria at 29,396,300 elements/ml, leukocyturia at 754,500 elements/ml and a negative culture. A non-regenerative normochromic normocytic anaemia at 9.1 g/dl, without hemostatic abnormalities, with the incidental discovery of severe renal failure (serum creatinine 654 µmol/L, urea 20.1 mmol/L). The presence of corrected hypocalcemia at 2.05 mmol/L, an elevated parathyroid hormone at 3.5 times the normal value and hypovitaminosis D at 20 ng/ml suggested the chronic nature of the renal failure. Urinary sediment analysis revealed glomerular-type proteinuria estimated at 2.07 g/24h, associated with hypoalbuminemia at 32 g/l.
A first-line etiological work-up was performed: serum protein electrophoresis as well as immunofixation were non-contributory; complement study, immunological work-up (ANCA and ANA) and viral serologies (HBV, HCV and HIV) were negative.
Radiological investigation
Imaging investigation of the hematuria led to an abdomino-pelvic CT scan. This revealed two kidneys in a lumbar position, enlarged, measuring 17 cm on the long axis bilaterally, with irregular contours containing multiple cystic lesions predominantly in the right kidney. Some lesions showed exophytic development with destruction of the renal parenchyma. The largest, measuring 8.1 cm in diameter, was located at the right lower pole. Several cysts had a hemorrhagic and fatty component. A right intra-pyelic hematoma measuring 80x81mm was also noted, appearing to communicate with the ipsilateral inferior polar artery, which showed aneurysmal dilatation. Examination of the abdomen also revealed a rounded hepatic cystic lesion of segment VIII, of mixed fatty and liquid density suggestive of an angiomyolipoma.
Diagnostic hypothesis
At this stage, given the association of renal cystic lesions and fibro-angiomatous cutaneous manifestations, the picture was strongly suggestive of Tuberous Sclerosis Complex complicated by chronic renal failure.
A contiguous gene syndrome, in particular a deletion involving the PKD1 gene, was also considered, given the bilateral nephromegaly and the presence of associated hepatic cysts.
Extension work-up
Given the suspicion of TSC, an extension work-up was performed and revealed several extra-renal locations:
Persistent pyrosis and hiccups, possibly related to phrenic compression; a complementary gastroduodenal fibroscopy was performed showing atrophic antro-fundic gastritis of metaplastic appearance on histopathology, with chronic interstitial duodenitis, mild chronic antritis and the presence of Helicobacter pylori.
Cardiac investigation by transthoracic echocardiography showed a preserved ejection fraction of 60% with a large right intraventricular mass suggestive of an intracavitary rhabdomyoma.
Ophthalmological examination found visual acuity at 7/10 OD and 10/10 OS; examination of the anterior segment was unremarkable. The fundus showed a budding papillary formation with vascular displacement, suggestive of an intra-retinal astrocytoma (Figure 3). A complementary cerebro-orbital MRI was indicated and was in favour of an astrocytic hamartoma of the retina given the context of Tuberous Sclerosis Complex.
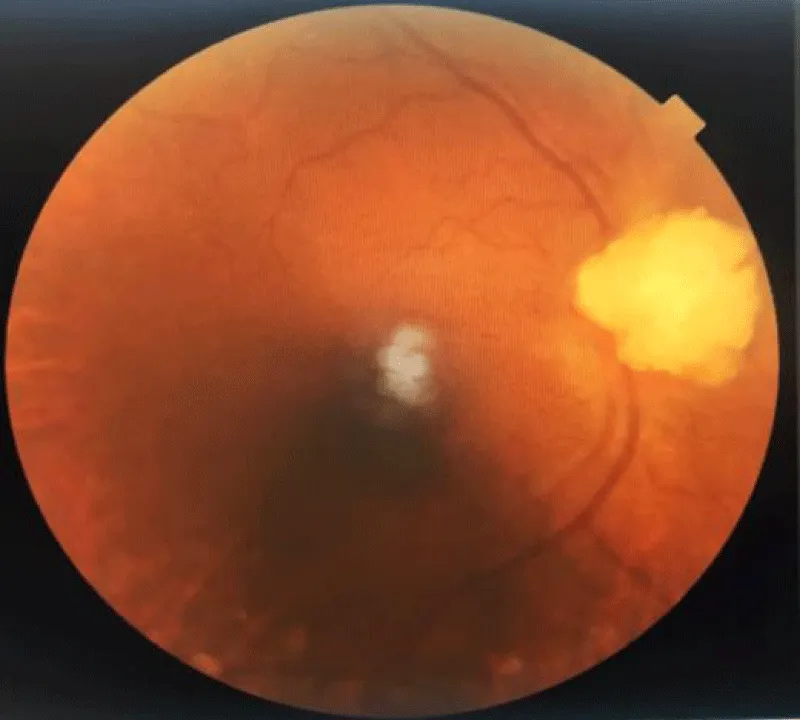
Figure 3: Fundus showing an intra-retinal astrocytoma observed in case 1.
On cerebral imaging, calcified subependymal nodular lesions were found in both lateral ventricles, without associated cortical tubers, with the additional presence of a right cerebellar calcification of 16 mm in diameter (Figure 4).
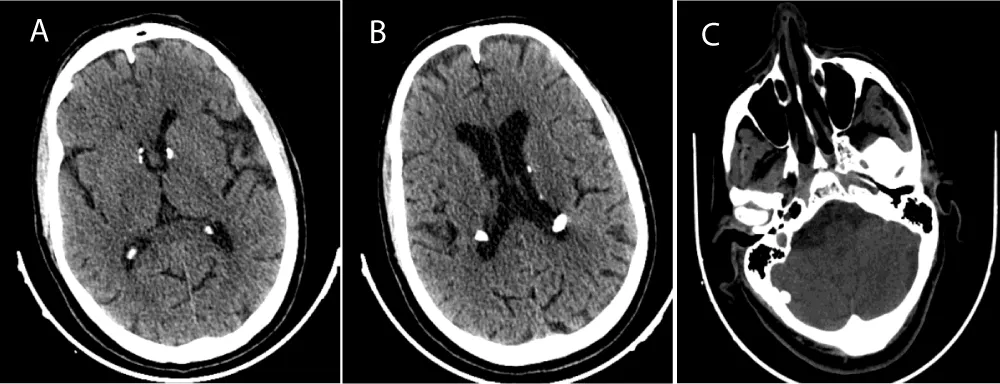
Figure 4: Axial CT slices of the cerebral level showing calcified subependymal nodular lesions in both lateral ventricles (A-B), with a right cerebellar calcification C (case 1).
Management
Therapeutically, in addition to the hemostatic treatment, the patient underwent selective radiological embolisation of the right inferior polar artery using biological glue. The CT scan check-up on day 3 was satisfactory, showing stabilisation of the lesions with regression of the hematoma dimensions (82×77 mm) (Figure 5).
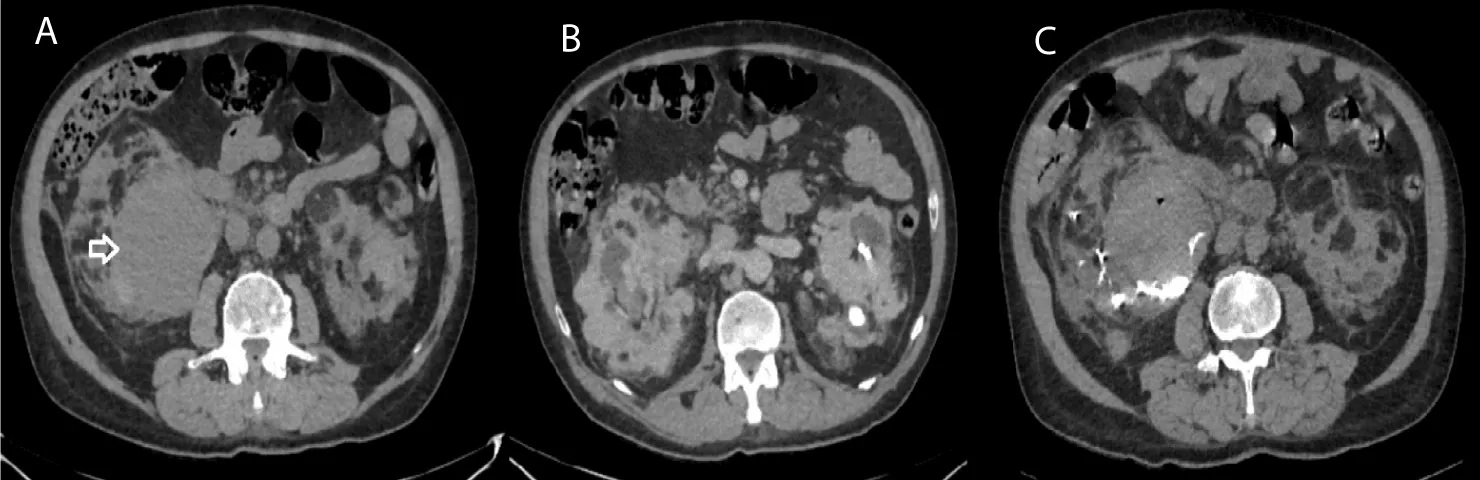
Figure 5: Axial CT acquisitions without iodinated contrast injection showing the parapyelic hematoma before and after selective arterial embolisation (case 1). A. Large right parapyelic renal cyst, with spontaneously hyperdense content, related to an intracystic bleeding (White Arrow) B. Bilateral renal parenchymal lesions with exophytic development for some, rounded and well-defined, heterogeneous, containing a fatty and tissue component. C. Radiological check-up after selective arterial embolisation of the right inferior polar artery with biological glue
Evolution
Renal evolution was marked by progression to end-stage renal disease requiring chronic hemodialysis.
A familial screening was undertaken, revealing cutaneous involvement characterised by hypopigmented macules and angiofibromas in four sons and two daughters. Renal involvement, characterised by the presence of bilateral renal cysts, was also found in one son and two daughters (Figure 1).
Genetic analysis was not performed.
Genotype–phenotype note: in the absence of molecular testing, the confirmed autosomal-dominant familial transmission with extensive multi-organ involvement (renal, cardiac, retinal, cerebral) but preserved neurocognition does not allow a reliable TSC1-versus-TSC2 prediction; genetic confirmation would be required.
Patient MZ, 45 years old, married without children. Referred from the dermatology clinic for the management of a patient already diagnosed as a carrier of Tuberous Sclerosis Complex since the age of 25, not genetically confirmed (Table 2a and 2b).
| Table 2a: Case 2 – summary against the 2021 international TSC diagnostic criteria. | |
| Major criteria present | Minor criteria present |
| Facial angiofibromas; cortical tubers; subependymal nodules (≥2); lymphangioleiomyomatosis; renal AML (≥2) | Multiple renal cysts; non-renal hamartoma (bone hamartoma) |
| ≥2 major criteria → clinically definite TSC (genetically unconfirmed). | |
| Table 2b: Case 2 – clinical timeline. | |
| Time | Event |
| ≈200(age 25) | TSC diagnosed clinically (not genetically confirmed); dermatology follow-up, Rabat |
| 2004 | Occipital scalp-tumour excision + skin-flap reconstruction |
| 2006 | Right nephrectomy for multicystic kidney; single left kidney thereafter |
| 2015 | Facial angiofibroma ablation (benign histology); CKD follow-up initiated |
| 2017 onwards | CHU Fès: repeated CO2-laser sessions + topical sirolimus 0.2% |
| 2015–2025 | Creatinine 115–159 µmol/L; current stable CKD G3b A1, eGFR 43 mL/min |
| Genetics | Not performed |
Past medical history
Chronic smoker for 20 years, 10 pack-years.
History of an occipital scalp tumour operated on in 2004 in the context of TSC, with reconstruction by skin flap.
Recurrent haemorrhoids with episodes of moderately abundant rectal bleeding reported in 2016.
Initially followed in dermatology at Souissi Hospital in Rabat between 2000 and 2012 for cutaneous involvement consisting of facial angiofibromas in the context of Tuberous Sclerosis Complex, then subsequently followed in dermatology at Fès University Hospital since 2017.
He underwent ablation of two facial angiofibroma lesions in 2015, the histopathological study of which concluded to a histological appearance compatible with a giant-cell angiofibroma without malignancy.
He underwent a right nephrectomy in 2006 in Rabat for a multicystic kidney related to TSC renal involvement, and since 2015, he has been followed for chronic renal failure on a single functional left kidney.
Clinical examination
The patient is in good general condition, apyretic, with blood pressure of 140/90 mmHg. Cardiopulmonary auscultation is unremarkable. Diuresis is preserved with a negative urine dipstick. There is no associated urological symptomatology nor extra-renal, respiratory or digestive manifestations. At the cutaneous level, there are multiple angiofibromas involving the face, particularly the nasolabial folds, the wings of the nose and the chin (Figure 6). Presence of several pigmented macules on the lower back and buttocks. The patient shows no signs of epilepsy or intellectual disability.
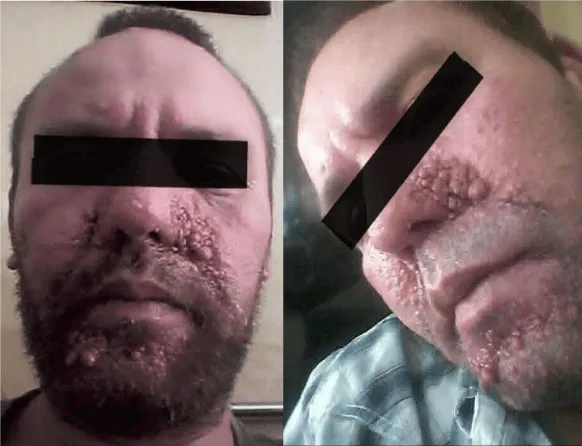
Figure 6: Multiple angiofibromas involving the face, on the nasolabial folds, the wings of the nose and the chin (case 2).
Laboratory work-up
On admission, a complete laboratory work-up confirmed the existence of renal failure with creatinine at 133 µmol/L and urea at 6.7 mmol/L without associated hydro-electrolyte disturbances.
The phosphocalcic balance was satisfactory, as well as a correct iron work-up without anaemia (Hb 16.2 g/dl). Metabolic, lipid and hepatic work-ups were normal, without glycemic disturbances (fasting glycemia at 0.86 g/l and HbA1C 5.5%) and PSA within normal limits.
24-hour proteinuria was negative. Urinary cytobacteriological examination showed neither leukocyturia nor hematuria.
Radiological investigation
Abdomino-renal ultrasound shows a homogeneous liver, of normal size, without dilatation of the intrahepatic (IHBD) or extrahepatic (EHBD) biliary ducts. The pancreas and spleen are normal. The right kidney is not visualised (previous nephrectomy); the left kidney measures 82 mm, appears slightly hyperechoic with poor corticomedullary differentiation. It is the site of multiple nodular formations, the largest of which measures 28 mm in diameter. There is also a simple cystic lesion of 21 mm in diameter. No dilatation of the pyelo-caliceal cavities. In conclusion: ultrasound appearance suggestive of left renal angiomyolipomas.
Dermatological management
Treatment consisted of the ablation of multiple facial angiofibromas through several sessions of ablative CO2 laser.
The patient underwent excision of a retro-auricular lesion compatible with a fibrous papule, a lesion of the left lower eyelid and a nodule at the inner canthus of the right eye, all compatible with an epidermoid cyst on histopathological study.
He also underwent surgical laser ablation of two flesh-colored papules on the back, showing fusocellular proliferation whose nature remains to be determined by immunohistochemical study.
Treatment with Sirolimus (mTOR inhibitor) cream was initiated for the angiofibromas.
Extension work-up
As part of the extension work-up, a thoraco-abdomino-pelvic CT scan (TAP) with iodinated contrast injection was performed. At the cerebral level, it revealed calcified subependymal nodules in both lateral ventricles, associated with cortical tubers. At the thoracic level, the images were in favour of lymphangioleiomyomatosis. Abdominal exploration confirmed the presence of multiple and bilateral renal angiomyolipomas associated with renal cysts. In addition, spinal analysis showed a bone hamartoma, while pelvic study revealed bone lesions of dysplastic appearance (Figure 7).
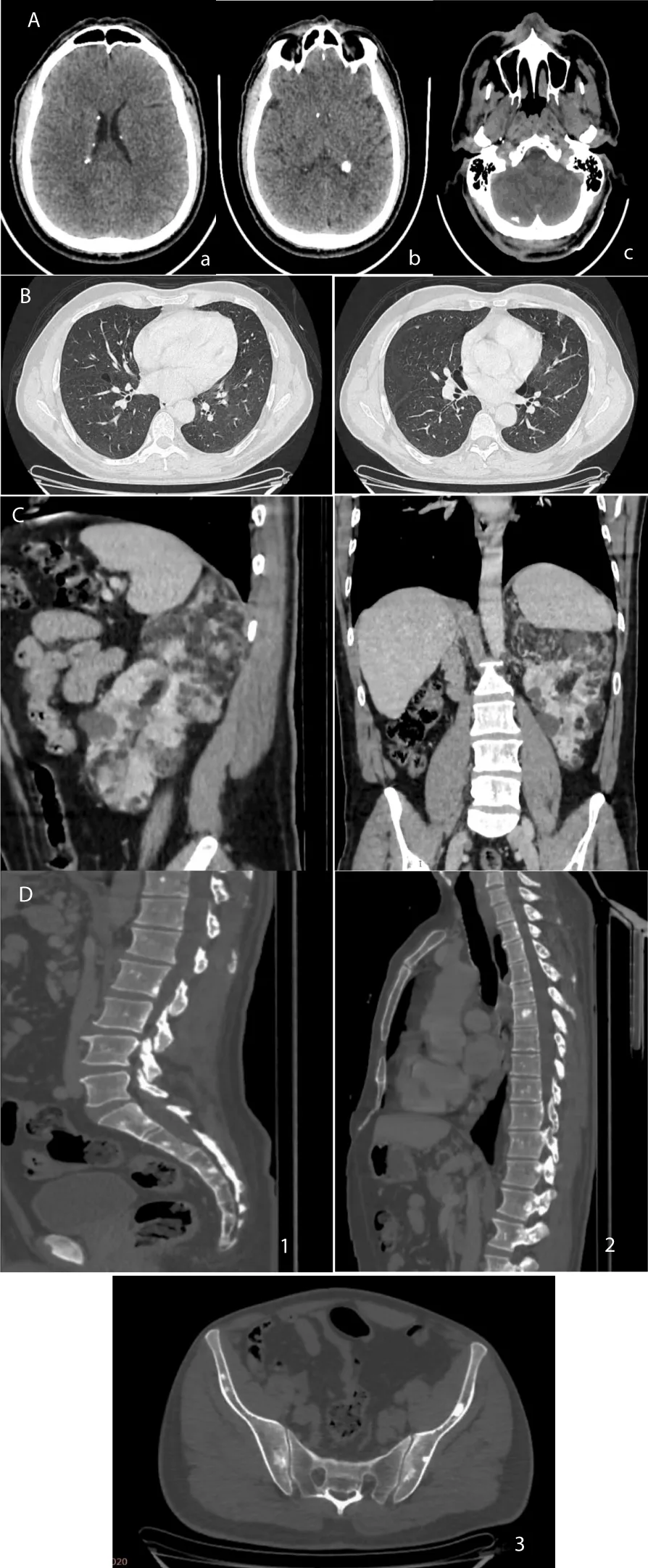
Figure 7: CT images of the various involvements of the patient (case 2). A: Axial slices through the cerebral level on non-enhanced acquisition showing calcified subependymal nodules (a-b) and right hemicerebellar calcifications suggestive of a cortical tuber c. B: Axial slices through the thoracic level in parenchymal window showing some thin-walled cystic lesions suggestive of lymphangioleiomyomatosis. C: Sagittal, coronal and axial slices through the abdominal level showing multiple angiomyolipomas and renal cysts. D: Sagittal and axial slices in bone window through the lumbosacral spine (1), dorsolumbar (2), and the pelvis (3) showing osteosclerotic lesions and areas of the vertebral bodies and spinous processes of the dorsolumbosacral spine as well as the iliac wings and sacrum..
In addition, ophthalmological examination with fundus showed no ocular involvement. Transthoracic echocardio-graphy was also unremarkable.
Evolution
On the renal level, the patient’s creatinine fluctuated between 115 and 159 µmol/L over ten years (2015–2025).
Currently, the patient presents with stable chronic renal failure, classified as stage G3b A1, with a baseline serum creatinine of 159 µmol/L and an estimated glomerular filtration rate of 43 ml/min.
In addition, he benefits from regular dermatological follow-up, with laser sessions, which have led to clear clinical improvement.
Topical sirolimus was added to the CO2-laser programme for the facial angiofibromas. Systemic mTOR inhibitor therapy (e.g., everolimus) was deliberately not initiated: current recommendations reserve systemic mTOR inhibition for renal angiomyolipomas that are ≥3 cm and/or growing, symptomatic, or at high haemorrhagic risk (size >4 cm or intratumoural aneurysm >5 mm), based on the EXIST-2 trial and the 2021 international guidelines. In this patient, the angiomyolipomas on the single remaining kidney were all < 3 cm (largest 28 mm), stable and without aneurysm, and renal function was stable; the balance between an indefinite, only partially cytoreductive treatment carrying recognised systemic toxicity and a low bleeding risk did not favour systemic therapy. Systemic mTOR inhibition was therefore reserved for any future AML progression.
Genotype–phenotype note: the sporadic presentation (no family history) with pulmonary lymphangioleiomyomatosis and a comparatively indolent renal course is compatible with either genotype; LAM is more frequently associated with TSC2, but genotyping was not available.
Patient WR, 14 years old, the second of a sibship of four boys (Figure 8), born to non-consanguineous parents, admitted to the pediatric emergency department for facial oedema associated with headaches and bilateral decreased visual acuity, evolving in a context of two episodes of mild epistaxis (Table 3a and 3b).
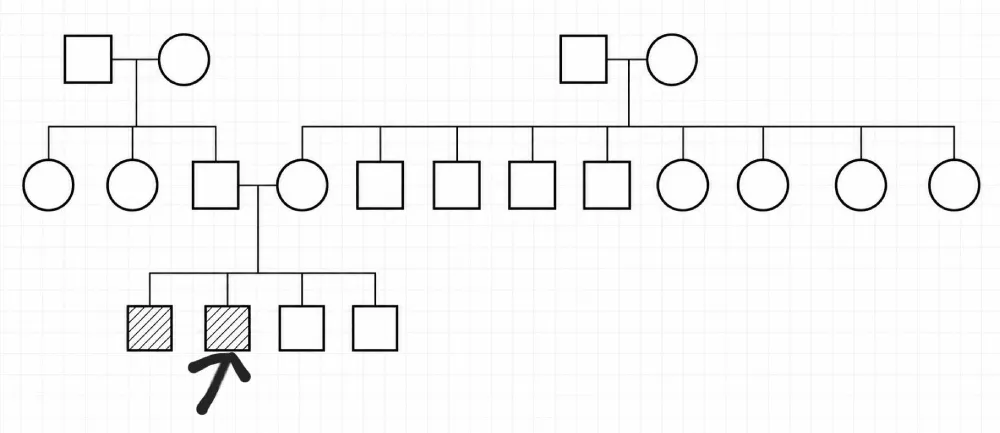
Figure 8: Family tree showing the screening in the family of case 3 (case 2). Legend: Arrow: designates the patient; Square: Male; Circle: Female; Crossed-out symbol: Deceased person: Empty symbol: Healthy person Hatched symbol: Affected person Horizontal line between two persons: Marriage / Union ┊┊┊ Vertical descending line: Descendants (children).
Table 3a: Case 3 – summary against the 2021 international TSC diagnostic criteria. |
|
Major criteria present |
Minor criteria present |
Hypomelanotic macules (≥3, ≥5 mm); retinal astrocytic hamartoma |
Multiple bilateral renal cysts |
2 major criteria → clinically definite TSC (genetically unconfirmed); no cerebral or cardiac involvement on imaging. |
|
| Table 3b:Case 3 – clinical timeline. | |
| Time | Event |
| Childhood | Recurrent tonsillitis; no prior TSC work-up |
| Admission (age 14) | Hypertensive emergency 170/110 mmHg; facial and lower-limb oedema; bilateral decreased visual acuity; epistaxis; advanced CKD (creatinine 707 µmol/L) with nephrotic-range proteinuria |
| Diagnosis | Retinal astrocytic hamartoma + hypomelanotic macules → TSC; bilateral cortical renal cysts + caudal pancreatic cyst; LV hypertrophy |
| Acute treatment | Antihypertensives + diuretic → worsening → methylprednisolone pulses + emergency haemodialysis + RBC transfusion; renal biopsy: chronic glomerulonephritis (7/7 globally sclerosed glomeruli) |
| Follow-up | Chronic haemodialysis since age 14; familial screening: 3 brothers normal, eldest brother → CKD → haemodialysis 8 years later |
| Current (age 28) | Preparing for cadaveric renal transplantation |
| Genetics | Not performed |
Past medical history: Recurrent tonsillitis during childhood.
Clinical examination
On admiOn examination, the patient was conscious, apyretic, with severe arterial hypertension at 170/110 mmHg.
Clinical examination revealed a puffy face with palpebral oedema as well as significant oedema of the lower limbs.
Diuresis was preserved. The urine dipstick showed proteinuria at two crosses, without hematuria.
Laboratory work-up
The renal work-up revealed advanced renal failure (serum creatinine at 707 µmol/L; urea 25 mmol/L) associated with hydro-electrolyte disturbances with dilutional hyponatremia at 125 mEq/l but without hyperkalemia.
The phosphocalcic work-up showed hypocalcemia at 2.0 mmol/L, hypoalbuminemia at 28 g/l, hyperphosphoremia at 2.42 mmol/L and parathyroid hormone at 13 times the normal value without vitamin D deficiency.
The haematological work-up showed a normochromic normocytic anaemia at 7.6 g/dL with serum ferritin at 500 µg/l.
Radiological investigation
Renal ultrasound showed small kidneys with the site of multiple bilateral renal cysts without dilatation of the excretory tract.
Extension work-up
Dermatological examination revealed cutaneous hypopigmented macules with café-au-lait spots.
The fundus revealed an astrocytic hamartoma.
Transthoracic echocardiography revealed left ventricular hypertrophy with preserved systolic function, associated with elevated filling pressures, without pulmonary arterial hypertension. The presence of a pericardial detachment was also noted.
Cerebral and thoracic CT scans showed no involvement related to Tuberous Sclerosis Complex.
The abdominopelvic CT scan revealed multiple bilateral renal cortical cysts associated with a caudal pancreatic cyst, without signs of malignancy on MRI (Figure 9).
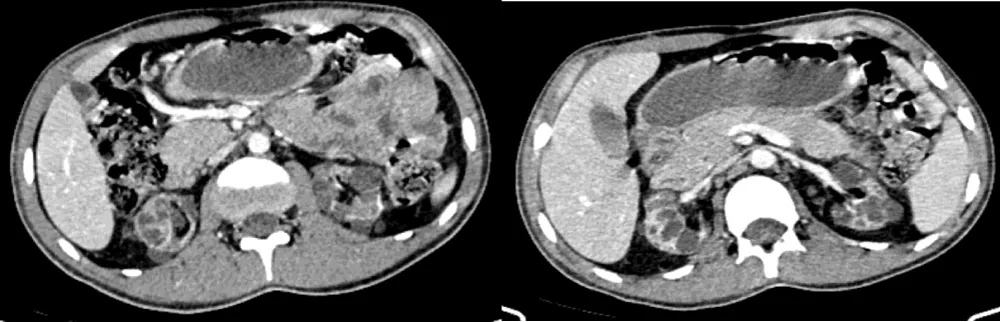
Figure 9: Axial CT slices of the abdominal level with iodinated contrast injection showing multiple bilateral renal cysts. (case 3)
A fibre colonoscopy was performed because of persistent diarrhoea and showed no significant abnormalities.
Nephrological management
Initially, the patient was started on antihypertensive treatment associated with a diuretic, allowing clinical improvement. However, a biological worsening of renal function was observed, prompting administration of Solu-Medrol boluses and emergency hemodialysis sessions with packed red blood cell transfusion, given the asymptomatic hyperuricemia and the threatening hyperkalemia, as well as management of complications related to chronic kidney disease.
In this context, a renal biopsy was performed and found seven totally fibrosed glomeruli, concluding chronic glomerulonephritis.
Evolution
Since his hospitalisation, the patient has been placed on chronic hemodialysis, started at the age of 14.
Now 28 years old, the patient is currently preparing his file for renal transplantation from a cadaveric donor.
Familial screening
Dermatological and ophthalmological examinations revealed no abnormalities in the three brothers. Regarding renal function, an alteration was found only in the eldest brother. Renal ultrasound showed small kidneys (7 cm) with lipomatous hyperechogenicity. He underwent a renal biopsy, which was in favour of chronic glomerulonephritis, and he was then started on chronic hemodialysis eight years after his brother (the patient) (Figure 8).
Genetic study was not performed
Genotype–phenotype note: the early, severe, cyst-predominant renal disease progressing to childhood end-stage renal disease – together with an affected sibling – would classically raise suspicion of a TSC2 variant, possibly with a contiguous TSC2/PKD1 deletion; genetic testing including PKD1 is recommended.
Interpretation of renal failure in case 3
The cause of end-stage renal disease in Case 3 is not fully explained by TSC-related cystic disease alone. The nephrotic-range presentation (heavy proteinuria, hypoalbuminaemia, oedema) with severe hypertension and a biopsy showing global glomerulosclerosis (7/7 sclerosed glomeruli, chronic glomerulonephritis) indicates a superimposed chronic glomerulopathy rather than isolated cystic nephropathy.
The main differential diagnoses are hypertensive nephroangiosclerosis, a primary chronic glomerulonephritis, and – given the concordant chronic glomerulonephritis in the elder brother – a hereditary glomerular or cystic nephropathy coexisting with, or independent of, TSC (e.g., a TSC2/PKD1 contiguous-gene syndrome). Genetic testing, including PKD1, would help clarify the underlying mechanism.
Diagnostic challenges
Several diagnostic challenges were common to all three cases. Genetic testing — the gold standard for TSC confirmation — could not be performed in any patient due to limited access to molecular genetics facilities and the high cost of high-throughput sequencing in our institutional setting. The diagnosis therefore relied exclusively on clinical and radiological criteria, in accordance with the 2021 revised international recommendations [11].
The phenotypic overlap of TSC with other cystic renal diseases — notably autosomal dominant polycystic kidney disease and PKD1 contiguous gene syndrome, formally considered as a differential in Case 1 — required comprehensive multidisciplinary evaluation. In Case 3, the severity of renal failure at presentation in a paediatric patient without prior TSC diagnosis posed additional diagnostic challenges.
Tuberous Sclerosis Complex (TSC) is an inherited multisystem disease, mainly affecting the central nervous system, skin, eyes and kidneys. It results from a germline mutation in the TSC 1 and TSC 2 genes, known as tumour suppressor genes, coding respectively for Hamartin and Tuberin. These two proteins negatively regulate the mTOR (mechanistic Target Of Rapamycin) pathway, which is involved in the regulation of cell growth and proliferation [1].
The incidence of TSC remains largely underestimated due to the rarity of recorded cases. Its current frequency is estimated at one case per 12,000 to 14,000 births in children under 10 years of age in the United Kingdom. It affects 1/20,000 to 1/25,000 births, respectively, in Europe and the United States [2]. There is no apparent sex or ethnic predominance [3,4].
Etiopathogenesis
As previously described, the mTOR (mechanistic Target Of Rapamycin) pathway is constitutively hyperactive in patients with TSC, resulting in hamartomatous formations in various organs. The TSC spectrum can occur either as familial forms (accounting for approximately 1/3 of cases) by autosomal dominant transmission, or in a sporadic mode, reflecting spontaneously occurring forms (2/3 of cases). Approximately 1–2% of familial forms may show germline mosaicism, explaining the absence of mutations in ascendants [4,5].
The TSC1 gene, located on chromosome 9 (q34.13), consists of 23 exons and codes for Hamartin (200 kDa) (Figure 10). Hamartin has many functional domains: an N-terminal portion (Exons 4–8), a domain of interaction with Tuberin (Exons 17–21), a domain interacting with the TBCD7 protein (Exons 22–23), and a final one with the HSP90 protein (Exon 23) (Figure 11). The variants determining the disease expression involve all coding parts of the gene, except exon 23 [4,6]. Reported TSC1 variants are predominantly truncating (nonsense mutations and deletions). TSC1 gene mutations account for 20–25% of mutations found in the disease and are associated with an often less severe phenotype [5,7,8].
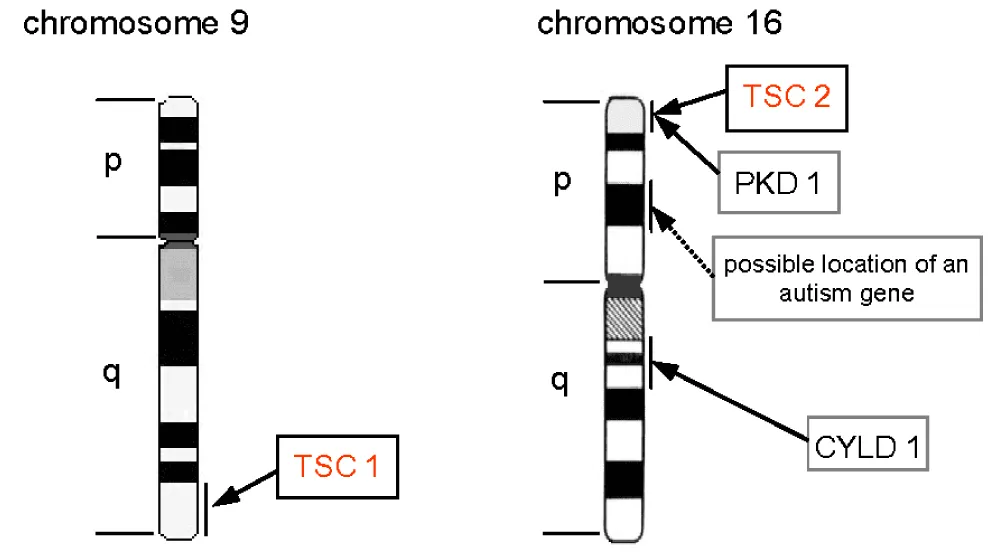
Figure 10: Diagram representing the location of the TSC1 and TSC2 genes on chromosomes 9 and 16, respectively [25].
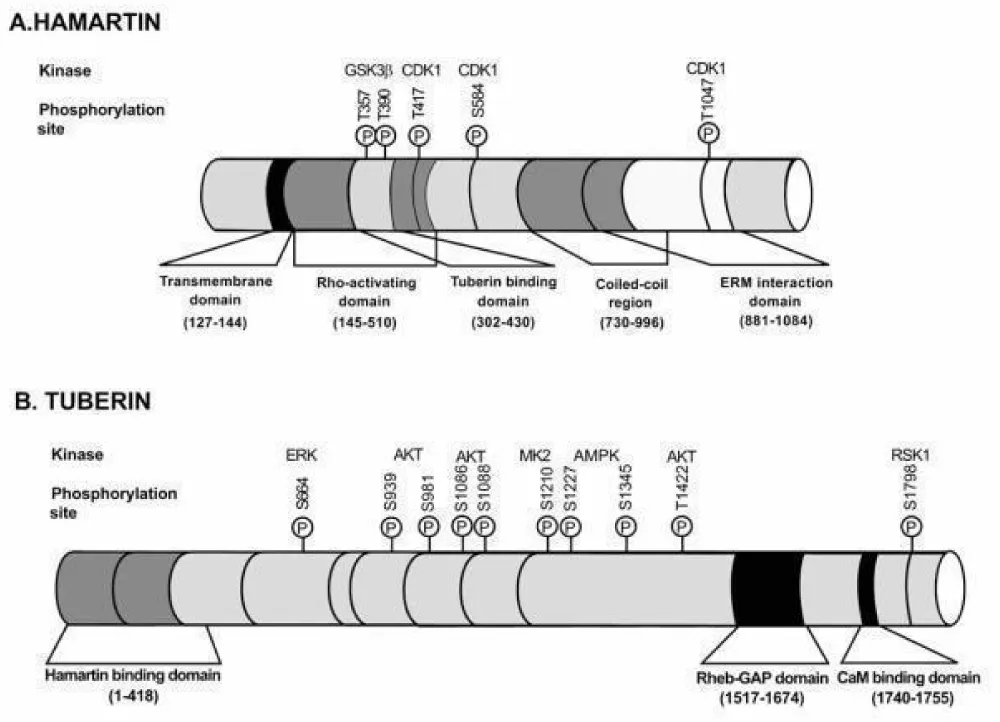
Figure 11: Schematic representation of the functional domains of Hamartin and Tuberin [26].
The TSC2 gene, located on chromosome 16p13, is composed of 23 exons coding for Tuberin (130 kDa) (Figure 10). It has an N-terminal domain (Exons 1–23), as well as a GTPase activating domain (Exons 35–40) [4,5] (Figure 11). Unlike the mutations found in TSC 1, TSC2 variants are more often deletions and missense mutations [7,10]. Two per cent of patients with a TSC2 mutation present a concomitant deletion of the PKD1 gene, resulting in a more severe phenotype combining that of TSC and autosomal dominant polycystic kidney disease.
Hamartin and Tuberin proteins are ubiquitously expressed. In association with the TBC1D7 protein, they form a heterotrimeric complex which negatively regulates the mTOR pathway. The action of the latter occurs through two protein complexes: mTOR 1 and 2 (Figure 12). The first is activated by the binding of activated RHEB to GTP. The inactivation of RHEB-GTP involves TSC2 via its GAP interaction domain. The expression of the disease is not entirely attributable to mTOR1, but would be due to a combined action of the activation of both proteins (mTOR1 and 2), via other mechanisms not yet elucidated [5,6].
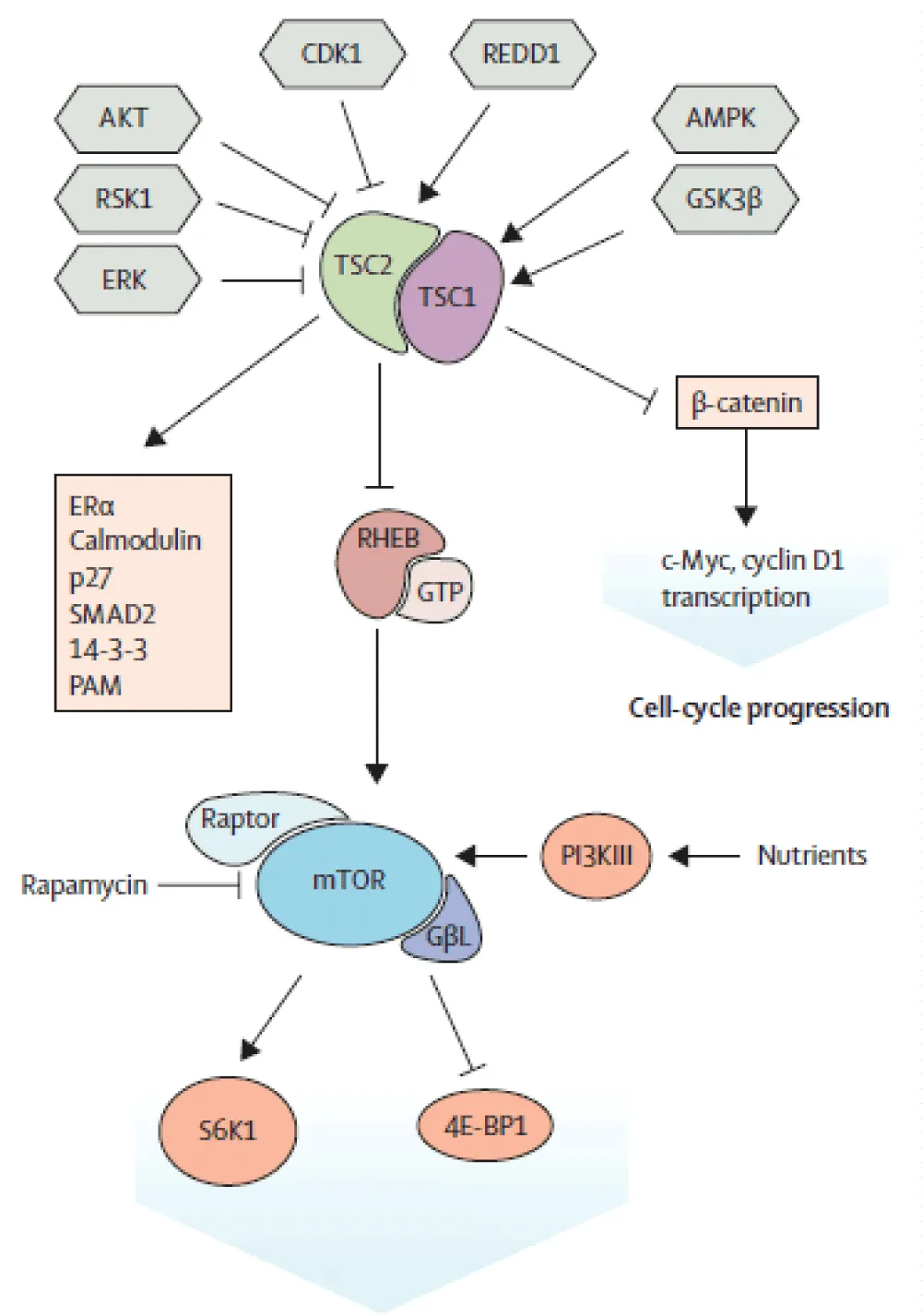
Figure 12: Molecular mechanisms of the mTOR signalling pathway [27].
Diagnostic criteria
The clinical manifestations of TSC are mainly related to the development of hamartomatous lesions, which exert an effect on adjacent healthy functional structures, giving rise to disease symptoms. Although rare, malignant transformations are providers of significant morbidity and mortality [10]. TSC preferentially affects the central nervous system, the heart, kidneys, skin, and more rarely the oral mucosa, bone structures and the digestive system. The most frequently encountered clinical mode of presentation in adults occurs either through the incidental discovery of renal involvement or on renal imaging, or on the occasion of complications (severe hypertension, retroperitoneal bleeding, or CKD), or through the discovery of lymphangiomyomatosis (LAM) (work-up of chronic dyspnea in a woman, recurrent pneumothorax), of cutaneous involvement (initially considered isolated), or more rarely of newly-onset epilepsy. In children, cutaneous and neurological manifestations are more frequent [3].
The diagnosis of the disease therefore relies on clinical, radiological, and sometimes histopathological and genetic criteria, and is the subject of international recommendations recently revisited in 2021 (Table 4). The diagnosis is made in the presence of either two major criteria, or one major criterion associated with at least two minor criteria. It remains possible in the presence of one major criterion and at least one minor criterion [11].
| Table 4:TSC diagnostic criteria according to the 2021 revised recommendations [11]. | |
| Major criteria | Minor criteria |
| Hypochromic macules (⩾3, at least 5 mm) “ash-leaf spots” | “Confetti” hypopigmented macules |
| Cephalic cutaneous fibrous plaque | Dental enamel pits (⩾3) |
| Ungual fibroma (⩾2) | Non-renal hamartoma |
| Shagreen patches (skin) | Multiple renal cysts |
| Multiple retinal hamartomas | Retinal achromic patch |
| Cortical tubers | Gingival fibromas (⩾2) |
| Subependymal nodules (⩾2) | |
| Subependymal giant-cell astrocytoma | |
| Cardiac rhabdomyoma | |
| Lymphangiomyomatosis (LAM) | |
| Renal angiomyolipoma (⩾2) | |
The yield of molecular diagnosis, although contributory even in the absence of clinical signs, is highly dependent on the technique used. With conventional molecular analyses, genetic analysis is negative in 15% of patients. High-throughput sequencing remains the reference technique, allowing identification even of mosaic forms [12,13].
Manifestations of TSC
Angiomyolipomas (AML) and cystic formations are the entities most often encountered at the renal level. Other tumours such as oncocytomas and renal carcinomas have been reported but remain less frequent, particularly in children [14]. Angiomyolipomas are hamartomatous formations characterised by the presence of three components: vascular, adipose and muscular. They are thought to derive from perivascular epithelioid cells, presenting an angiogenic potential and lipid accumulation [15]. These tumours often appear early, around the age of 10, and are encountered in 80% of cases in adults [11]. Cystic formations are often bilateral and multiple, and are found in 30 to 45% of cases. AMLs are often asymptomatic and more rarely discovered during certain complications: hypertension, lumbar pain, renal failure or hematuria. The rate of spontaneous hemorrhagic rupture is 5 to 6% per year [16,17]. Tumour size (> 4 cm) [11], the presence of an aneurysm of more than 5 mm [18], and estrogen exposure [19] are all factors implicated in the occurrence of hemorrhagic complications.
Renal carcinomas are rare, affecting approximately 2 to 5% of patients, but remain more frequent than in the general population [3]. The mean age at diagnosis is 30 years; however, they can present at an earlier age. The most frequent histological subtype is clear-cell carcinoma. These are mainly suspected in the presence of a rapidly growing renal mass without a fatty component, with calcifications or a central necrotic area. Learned societies recommend surveillance every 1 to 3 years, preferably by MRI [11].
Genotype-phenotype correlation
It is clinically impossible to distinguish between TSC 1 and TSC 2 forms; however, many studies conclude to a more severe phenotype in TSC 2 forms [20]. The latter seems to expose patients to an increased risk of intellectual disability, cancers, infantile spasms and LAM.
The distribution of symptoms among individuals carrying the same mutation remains rather heterogeneous (Figure 13). This disparity can be explained by the so-called “Second Hit” theory, which involves an additional mutation on the healthy allele of the TSC mutation, thereby implying differentiation of a certain group of cells towards the development of hamartomas (Figure 14). This theory therefore implies a loss of heterozygosity, which explains the variability of the clinical expression of the disease.
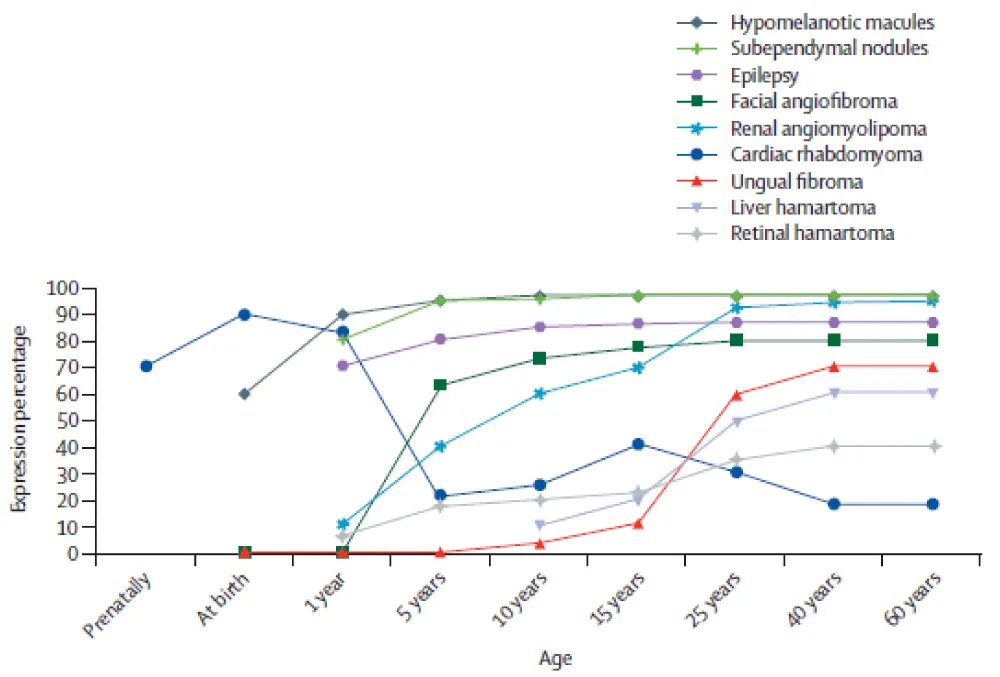
Figure 13: Distribution of clinical manifestations of Tuberous Sclerosis Complex according to age [27].
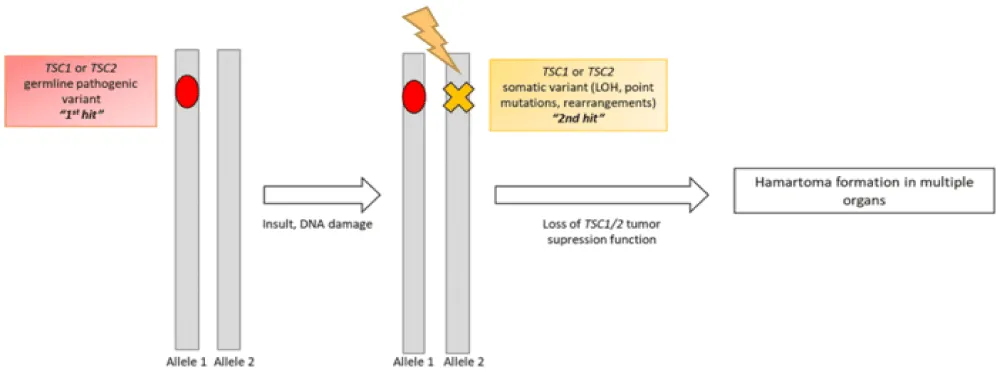
Figure 14: “Second Hit” hypothesis applied to Tuberous Sclerosis Complex [28].
Practical nephrological implications
These cases underline four priorities for the nephrologist. First, systematic renal imaging at diagnosis and lifelong surveillance of angiomyolipomas and cysts – by MRI where available – every 1 to 3 years. Second, active renal preservation: strict blood-pressure control, preference for selective arterial embolisation over nephrectomy for bleeding or high-risk AMLs, and timely systemic mTOR inhibition for AMLs ≥3 cm or growing. Third, systematic familial screening, which disclosed additional affected relatives in two of our three families. Fourth, lifelong multidisciplinary follow-up involving nephrology, dermatology, cardiology, ophthalmology, neurology, radiology and genetics.
Evolution and prognosis
The risk of progression to end-stage chronic renal failure is approximately 1% [21]. This risk seems to be conditioned by the size of the cysts and the number of AMLs, the use of surgery and selective arterial embolisation reducing the remaining functional renal parenchyma, and arterial hypertension, which constitutes a factor of progression of renal disease [14].
Surgical and medical therapeutic modalities
The therapeutic management of patients with TSC must be the subject of multidisciplinary consultations because of the multisystem nature of the disease. Thus, the management of renal AMLs in the context of TSC remains variable between teams, but remains correlated with the hemorrhagic risk and the size of the tumour. Arterial embolisation remains the most widely adopted technique in tumours with selective vascularisation whose size is > 4 cm, or those associated with aneurysmal lesions > 5 mm. It allows a reduction of the hemorrhagic risk, as well as preservation of renal function in the short and long term [22], but has no impact on AML growth and exposes to a risk of recurrence in case of interruption.
The advent of mTOR inhibitors is taking an increasing place in the treatment of AMLs, allowing not only a reduction in size but also a reduction in long-term hemorrhagic risk [23,24]. The disadvantage of these treatments is mainly related to side effects, but also to the fact that they only allow a reduction of the lesions without complete eradication.
Surgical treatment (total nephrectomy) constitutes the last-resort armada, mainly for hemostatic purposes in hemorrhagic situations related to a cyst rupture [11].
Strengths and limitations
This case series presents several strengths: it illustrates the full clinical spectrum of TSC through three patients with markedly distinct presentations (Table 5), each undergoing thorough multisystem evaluation per the 2021 international criteria. Familial screening in two cases revealed additional affected relatives, highlighting the practical value of systematic family investigation.
| Table 5: Clinical, renal, extra-renal and evolutionary characteristics of the three patients with TSC | |||
| Characteristics | Case 1 | Case 2 | Case 3 |
| Age / Sex | 65 years / M | 45 years / M | 14 years / M |
| Mode of revelation | Macroscopic hematuria | Known cutaneous involvement | Oedema + severe hypertension |
| Cutaneous involvement | Angiofibromas, ungual fibromas, shagreen patches | Facial angiofibromas | Hypopigmented macules |
| Neurological involvement | Absent | Absent | Absent |
| Renal involvement | AML + bilateral cysts | Left kidney AML + CKD | Bilateral cysts + end-stage CKD |
| Renal function | Severe CKD | Stable moderate CKD | End-stage CKD |
| Extra-renal involvement | Retinal astrocytoma, cardiac rhabdomyoma | Pulmonary LAM, bone lesions | Retinal hamartoma |
| Treatment | Embolization + hemodialysis | Laser + topical Sirolimus | Chronic hemodialysis |
| Form | Familial | Sporadic | Probable familial |
| Evolution | Chronic HD | Stable | Awaiting renal transplantation |
| AML: angiomyolipoma; CKD: chronic kidney disease; LAM: lymphangioleiomyomatosis; HD: hemodialysis. | |||
Several limitations must be acknowledged. The absence of genetic testing in all three patients precludes definitive molecular diagnosis and limits genotype-phenotype correlation, reflecting the constraints of our institutional setting. The retrospective design limits completeness of follow-up data. Renal biopsy was not performed in Cases 1 and 2. As a three-patient case series, generalisation is inherently limited.
TSC remains a rare condition whose morbidity and mortality are closely correlated with the clinical phenotype. Disease expression is heterogeneous, depending both on the type of mutation and on the pathophysiological mechanism of the “Second Hit” theory. Renal involvement, dominated by angiomyolipomas (AML) and cystic formations, is frequent. However, progression to hemorrhagic complications or malignant transformation remains exceptional. The renal prognosis is generally favourable, with infrequent progression to end-stage disease, but requires early screening and regular follow-up.
Management must be multidisciplinary, adapted to the clinical phenotype and functional impact. Targeted molecular therapies represent a major therapeutic advance, particularly on the renal level, although they do not allow definitive cure.
Patient perspective
Not applicable. Individual patient perspectives on treatments could not be formally collected at the time of manuscript preparation due to logistical constraints inherent to the retrospective nature of this case series.
Declarations
Ethics approval and consent to participate: Ethics approval was not required for this case report, as per the institutional policy of Hassan II University Hospital, Fes, Morocco, which exempts small case series from formal ethics committee review. All patients provided written informed consent for the use of their clinical data for scientific publication purposes.
Consent for publication
Written informed consent for publication was obtained from all patients included in this report. Patients who were minors at the time of clinical presentation had reached the age of majority at the time of manuscript preparation and provided their own written informed consent for publication. In addition, we have also obtained written informed consent from the parent of the patient who was a minor at the time of the clinical presentation.
All clinical images (facial and cutaneous photographs, fundus images and family pedigrees) were reviewed and fully anonymised; identifying facial features were masked, and no individual patient is identifiable from the figures.
Availability of data and materials
All data generated or analysed during this study are included in this published case report. De-identified data supporting the findings are available from the corresponding author upon reasonable request.
Authors’ contributions
RB conceptualised the study, collected clinical data, and drafted the manuscript. BAC and OK contributed to data collection and interpretation of radiological findings. YA and GEB contributed to the literature review and critical revision. NK and TSH supervised the study, provided critical intellectual input, and approved the final version. All authors read and approved the final manuscript.
Acknowledgement
The authors wish to thank the patients and their families for their trust and consent to publication. The authors also acknowledge the multidisciplinary teams of the Nephrology, Dermatology, Ophthalmology, Cardiology, and Radiology Departments of Hassan II University Hospital, Fes, Morocco.
- Marom D. Genetics of tuberous sclerosis complex: an update. Childs Nerv Syst. 2020;36:2489‑2496. Available from: https://doi.org/10.1007/s00381-020-04726-z
- O’Callaghan FJ, Shiell AW, Osborne JP, Martyn CN. Prevalence of tuberous sclerosis estimated by capture‑recapture analysis. Lancet. 1998;351:1490. Available from: https://doi.org/10.1016/s0140-6736(05)78872-3
- Protocole National de Diagnostic et de Soins (PNDS) – Sclérose tubéreuse de Bourneville. Reference Centre for Rare Epilepsies, Lille University Hospital and Necker Enfants Malades, APHP. September 2021.
- Peron A, Sing KA, Northrup H. Genetics, genomics, and genotype–phenotype correlations of TSC: insights for clinical practice. Am J Med Genet C Semin Med Genet. 2018;178C:281‑290. Available from: https://doi.org/10.1002/ajmg.c.31651
- Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016 May 26:2:16035. Available from: https://doi.org/10.1038/nrdp.2016.35
- McEneaney LJ, Tee AR. Finding a cure for tuberous sclerosis complex: from genetics through to targeted drug therapies. Adv Genet. 2019;103:91‑118. Available from: https://doi.org/10.1016/bs.adgen.2018.11.003
- Cheadle JP, Reeve PR, Sampson JR, Kwiatkowski DJ. Molecular genetic advances in tuberous sclerosis. Hum Genet. 2000;107:97‑114. Available from: https://doi.org/10.1007/s004390000348
- Schwartz RA, Fernandez G, Kotulska K, Jóźwiak S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189‑202. Available from: https://doi.org/10.1016/j.jaad.2007.05.004
- Samueli S, Abraham K, Dressler A, Groeppel G, Jonak C, Muehlebner A, et al. TSC‑Zentrum Wien tuberous sclerosis complex: new criteria for diagnostic work‑up and management. Wien Klin Wochenschr. 2015;127:619‑630. Available from: https://doi.org/10.1007/s00508-015-0758-y
- Tuberous Sclerosis Complex. Orphanet Handicap Encyclopedia. November 2015. Available from: www.orpha.net/data/patho/Han/Int/fr/LaScleroseTubereuseDeBourneville_FR_fr_HAN_ORPHA805.pdf
- Northrup H, Aronow ME, Bebin EM, Bissler J, Darling TN, de Vries PJ, et al. Updated international tuberous sclerosis complex diagnostic criteria and surveillance and management recommendations. Pediatr Neurol. 2021;123:50‑66. Available from: https://doi.org/10.1016/j.pediatrneurol.2021.07.011
- Nellist M, Brouwer RWW, Kockx CEM, van Veghel-Plandsoen M, Withagen-Hermans C, Prins-Bakker L, et al. Targeted next generation sequencing reveals previously unidentified TSC1 and TSC2 mutations. BMC Med Genet. 2015;16:10. Available from: https://doi.org/10.1186/s12881-015-0155-4
- Tyburczy ME, Dies KA, Glass J, Camposano S, Chekaluk Y, Thorner AR, et al. Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet. 2015;11(11):e1005637. Available from: https://doi.org/10.1371/journal.pgen.1005637
- Trnka P, Kennedy SE. Renal tumours in tuberous sclerosis complex. Pediatr Nephrol. 2021;36:1427‑1438. Available from: https://doi.org/10.1007/s00467-020-04775-1
- Lam HC, Nijmeh J, Henske EP. New developments in the genetics and pathogenesis of tumours in tuberous sclerosis complex. J Pathol. 2017;241:219‑225. Available from: https://doi.org/10.1002/path.4827
- Nelson CP, Sanda MG. Contemporary diagnosis and management of renal angiomyolipoma. J Urol. 2002;168(4 Pt 1):1315‑1325. Available from: https://doi.org/10.1016/s0022-5347(05)64440-0
- Van Baal JG, Smits NJ, Keeman JN, Lindhout D, Verhoef S. The evolution of renal angiomyolipomas in patients with tuberous sclerosis. J Urol. 1994;152(1):35‑38. Available from: https://doi.org/10.1016/s0022-5347(17)32809-4
- Yamakado K, Tanaka N, Nakagawa T, Kobayashi S, Yanagawa M, Takeda K. Renal angiomyolipoma: relationships between tumour size, aneurysm formation, and rupture. Radiology. 2002;225(1):78‑82. Available from: https://doi.org/10.1148/radiol.2251011477
- Lu Y, Liu X, Zhang E, Kopras EJ, Smith EP, Astreinidis A, et al. Estrogen activates pyruvate kinase M2 and increases the growth of TSC2‑deficient cells. PLoS One. 2020;15(2):e0228894. Available from: https://doi.org/10.1371/journal.pone.0228894
- Yates JR. Tuberous sclerosis. Eur J Hum Genet. 2006;14:1065‑1073.
- Eijkemans MJ, van der Wal W, Reijnders LJ, Roes KCB, van Waalwijk van Doorn-Khosrovani SB, et al. Long‑term follow‑up assessing renal angiomyolipoma treatment patterns, morbidity, and mortality: an observational study in tuberous sclerosis complex patients in the Netherlands. Am J Kidney Dis. 2015;66:638‑645. Available from: https://doi.org/10.1053/j.ajkd.2015.05.016
- Williams JM, Racadio JM, et al. Embolisation of renal angiomyolipomata in patients with tuberous sclerosis complex. Am J Kidney Dis. 2006;47:95‑102. Available from: https://doi.org/10.1053/j.ajkd.2005.09.028
- Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST‑2): a multicentre, randomised, double‑blind, placebo‑controlled trial. Lancet. 2013;381:817‑824. Available from: https://doi.org/10.1016/s0140-6736(12)61767-x
- Bissler JJ, Nomomura N, Budde K, Johnson ND, Donnelly LF, Bissler JJ. Everolimus long‑term use in patients with tuberous sclerosis complex: four‑year update of the EXIST‑2 study. PLoS One. 2017;12:e0180939.
- Of the variability of GTPase activating protein (GAP) related domain of the tuberous sclerosis 2 (TSC2) gene in TSC patients and healthy subjects. Inaugural dissertation. 2004. Available from: https://www.semanticscholar.org/paper/OF-THE-VARIABILITY-OF-THE-GTPase-ACTIVATING-GAP-)-(/e30f52bc5cd9e9c8b102a963cc793238ec1c984b
- Napolioni V, Curatolo P. Genetics and molecular biology of tuberous sclerosis complex. Curr Genomics. 2008;9(7):475‑487. Available from: https://doi.org/10.2174/138920208786241243
- Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657‑668. Available from: https://doi.org/10.1016/s0140-6736(08)61279-9
- Hino O, Kobayashi T. Mourning Dr Alfred G. Knudson: the two‑hit hypothesis, tumour suppressor genes, and the tuberous sclerosis complex. Cancer Sci. 2017;108(1):5‑11. Available from: https://doi.org/10.1111/cas.13116