
More Information
Submitted: December 07, 2023 | Approved: December 18, 2023 | Published: December 19, 2023
How to cite this article: Varanasi L, Loeb G, Walavalkar V, Mohammed N, Lindsey II JP, et al. Kidney Biopsy in Autosomal Dominant Polycystic Kidney Disease. J Clini Nephrol. 2023; 7: 102-105.
DOI: 10.29328/journal.jcn.1001118
Copyright License: © 2023 Varanasi L, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Polycystic kidney disease; Proteinuria; Kidney biopsy; Genetic testing

Kidney Biopsy in Autosomal Dominant Polycystic Kidney Disease
Laalasa Varanasi1*, Gabriel Loeb1, Vighnesh Walavalkar2, Nebil Mohammed3, John Paul Lindsey II4, Stephen Gluck5, Thomas Lee Chi6 and Meyeon Park7
1Assistant Professor, Medicine, University of California San Francisco, USA
2Associate Professor, Pathology, University of California San Francisco, USA
3Resident Physician, Pathology, University of California San Francisco, USA
4Resident Physician, Urology, University of California San Francisco, USA
5Professor, Medicine, University of California San Francisco, USA
6Professor, Urology, University of California San Francisco, USA
7Associate Professor, Medicine, University of California San Francisco, USA
*Address for Correspondence: Laalasa Varanasi, MD, Assistant Professor, Medicine, University of California San Francisco, USA, Email: [email protected]
Proteinuria is an easily quantified biomarker of kidney disease and often a sign of glomerular pathology. Significant proteinuria is uncommon in cystic kidney diseases and should be further evaluated to exclude the presence of another simultaneous kidney disease. While renal biopsy is a valuable part of the diagnostic evaluation of proteinuria, careful consideration of risks and benefits is necessary before proceeding in a patient with bilateral renal cysts. We report the case of a man with Polycystic Kidney Disease (PKD) who was found to have nephrotic-range proteinuria. An ultrasound-guided kidney biopsy revealed evidence of Focal Segmental Glomerulosclerosis (FSGS), which was attributed to hyperfiltration-related injury in the context of extensive kidney cysts. Genetic testing did not reveal a cause of FSGS and showed a variant of uncertain significance in PKD1. We use this case to highlight three important issues that are applicable to patients with PKD: the role of diagnostic evaluation for proteinuria in cystic kidney disease, the feasibility of kidney biopsy despite the presence of bilateral renal cysts, and the roles and limitations of genetic testing in cystic kidney disease and FSGS.
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is a common monogenic disease, which accounts for ~6% of kidney failure [1]. ADPKD is characterized by the development of bilateral kidney cysts and associated kidney enlargement; cysts are also seen in the liver, pancreas, and epididymis in significant fractions of patients. Although family history and imaging findings consistent with ADPKD can often establish the diagnosis of ADPKD, there are a number of other cystic diseases that should be considered in the differential diagnosis. Significant proteinuria is uncommon in ADPKD, with only about 27% of patients demonstrating greater than 300 milligrams per day [2]. The severity of microalbuminuria and proteinuria is highly variable, so experts have suggested that patients with ADPKD who have proteinuria of greater than 2 grams per day should undergo further evaluation to exclude the presence of another simultaneous kidney disease [2-4]. Among the few existing case reports describing ADPKD associated with nephrotic syndrome, focal segmental glomerulosclerosis was the most frequent diagnosis on renal biopsy [5].
We report the case of a man with a diagnosis of polycystic kidney disease and nephrotic-range proteinuria who underwent renal biopsy, which showed evidence of focal segmental glomerulosclerosis.
The patient is a man in his fifties with a history of hypertension, hypercholesterolemia, Crohn’s Disease, and type 2 diabetes mellitus without retinopathy who was referred to our nephrology clinic by his local nephrologist for a second opinion regarding the evaluation of proteinuria and consideration of tolvaptan for rapidly progressive ADPKD. Since his diagnosis of Crohn’s Disease in 2003, the patient had been on mesalamine and had multiple surveillance colonoscopies without evidence of malignancy. His other medications included amlodipine, metoprolol, losartan, hydralazine, atorvastatin, and calcium/vitamin D supplements. His father had multiple cysts in both kidneys and started dialysis at 76 years old. The patient began seeing a nephrologist in 2012 for proteinuria ranging from 1.5 to 2.0 grams per day. His estimated glomerular filtration rate (eGFR) was > 60 mL/min at the time. The following differential diagnoses were considered: hypertensive nephropathy, prolonged use of mesalamine, or AA amyloidosis in the setting of Crohn’s disease. Further work-up, including complement levels (C3 and C4), antinuclear antibody, anti-neutrophil cytoplasmic antibody, serum and urine protein electrophoresis, Hepatitis B and C, and HIV serologies, was negative. Renal ultrasound in January 2012 showed the right kidney measuring 12 cm, the left kidney measuring 13 cm, and multiple tiny, benign-appearing cysts bilaterally. Subsequently, a CT scan of the abdomen and pelvis in 2018 showed simple and complex renal cysts bilaterally and no cysts in the liver, spleen, or pancreas. Renal biopsy was initially deferred due to the presence of multiple renal cysts. The patient was eventually referred to our center for a second opinion regarding the management of polycystic kidney disease and proteinuria. At the time of referral in 2019, the physical exam was notable for high blood pressure of 144/86 but was otherwise normal without edema. The patient’s body mass index was 25 kg/m2. Laboratory testing revealed a serum creatinine of 2.09 mg/dL, eGFR of 35 mL/min, spot urinary protein to creatinine ratio of 5.1 g/g, and serum albumin of 3.5. Serum phospholipase A2 receptor (PLA2R) antibody testing was negative. Hemoglobin A1c was 6.2%. An MRI of the abdomen was performed and showed polycystic kidneys with a right kidney volume of 224 mL and a left kidney volume of 303 mL. The patient underwent an ultrasound-guided biopsy of the renal cortex, which was performed by a urologist in the operating room. Light microscopy (Figure 1a) revealed 12 glomeruli with Focal Segmental Glomerulosclerosis (FSGS), severe chronicity with 60% - 70% interstitial fibrosis and tubular atrophy, moderate global glomerulosclerosis, and moderate arteriosclerosis and arteriolosclerosis. There was no evidence of diabetic nephropathy, and immunofluorescence stains were negative. Electron microscopy revealed focally moderate effacement of podocyte foot processes (Figure 1b). The FSGS pattern of injury was thought to be secondary to hyperfiltration given the focality of the podocyte effacement and the underlying advanced kidney disease (Figure 2). Genetic testing (Natera Renasight panel) was notable for a heterozygous likely pathogenic variant in PKHD1 (pLys2003Serfs*30) and variants of unknown significance in PKHD1 (p.Gln2167Lys) and PKD1 (p.Arg1364His).
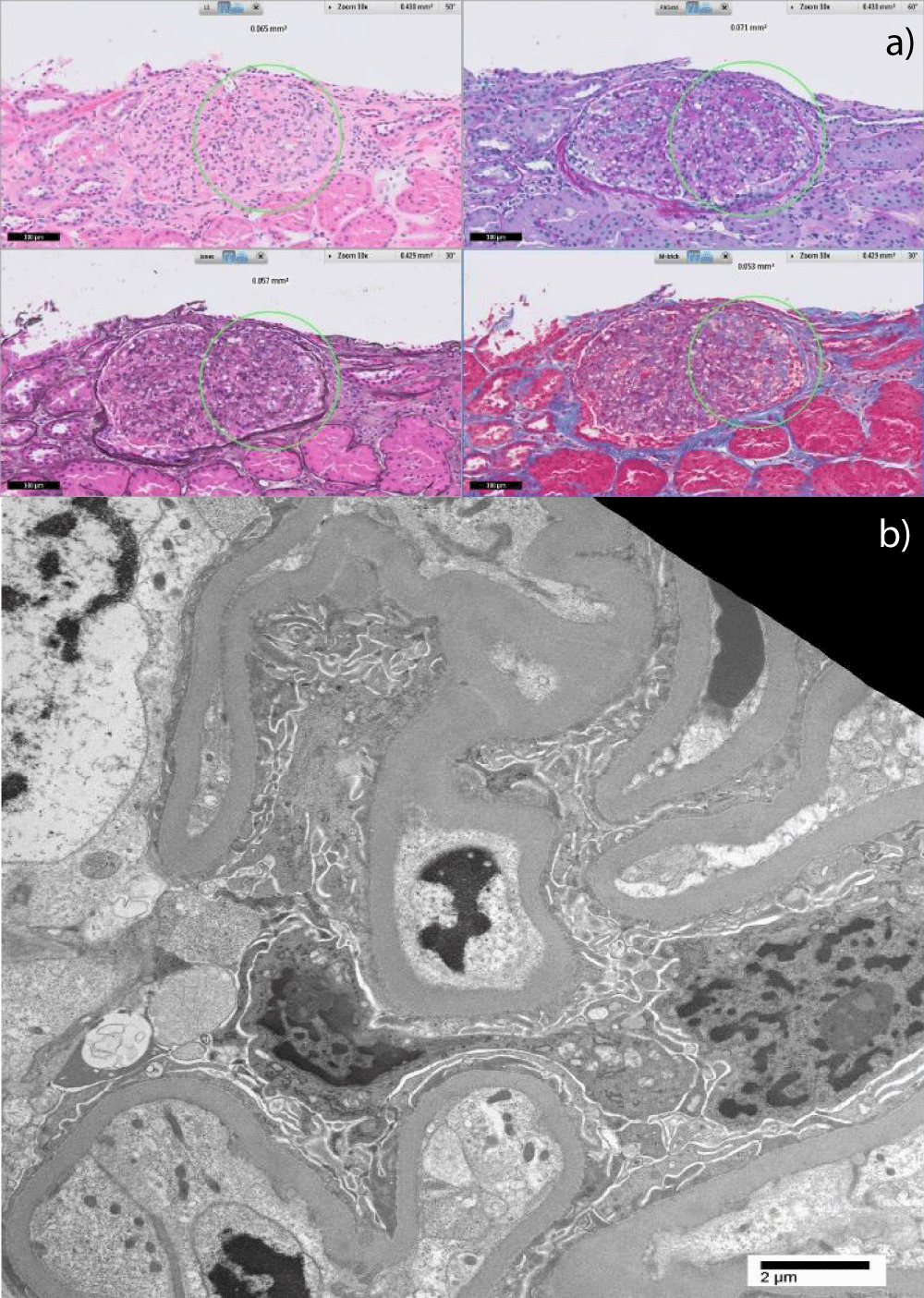
Figure 1: a) Light microscopy with various stains (clockwise starting at top left: hematoxylin & eosin, periodic acid Schiff, Masson’s trichrome, Jones methenamine silver) showing the perihilar variant of FSGS. The circled area highlights the consolidation of capillary loops with hyaline deposition and intracapillary foam cells. b) Electron microscopy showing moderate, not diffuse, effacement of podocyte foot processes suggestive of secondary FSGS.
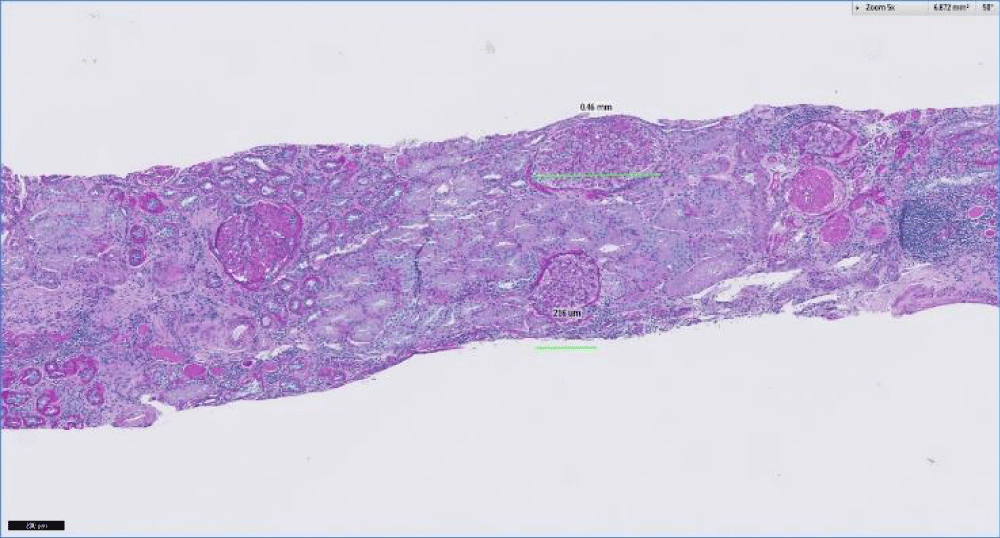
Figure 2: Light microscopy with Periodic acid-Schiff stain showing interstitial fibrosis, tubular atrophy, and enlarged glomeruli (> 250 nm) indicative of hyperfiltration.
Nephrotic range proteinuria is atypical in cystic kidney disease. In the Modification of Diet in Renal Disease Study (MDRD) [6], there were two hundred patients that had ADPKD, with mean urinary protein excretion of 0.29 g/day in patients with GFR of 25 - 55 mL/min/1.73 m2 and 0.46 g/day in patients with GFR of 13 - 24 mL/min/1.73m2. Nephrotic-range proteinuria or nephrotic syndrome in association with ADPKD is considered rare, and in our view, should be further investigated to exclude the presence of a superimposed glomerular disease. A review of the literature in 2012 by Visciano, et al. revealed that there were only 29 reported cases of ADPKD with nephrotic syndrome evaluated by renal histopathological studies. Among these reported cases there was a wide spectrum of glomerular disease including FSGS, minimal change disease, membranous nephropathy, and IgA nephropathy [5]. In a separate case report describing ADPKD accompanied by progressing FSGS, worsening glomerular segmental sclerosis, glomerular collapse, and progressive glomerular cyst formation, Oda, et al. hypothesized that glomerular hyperfiltration due to ADPKD caused glomerular cyst formation and development of FSGS [7]. In our case, hypertensive nephrosclerosis must also be considered in the differential diagnosis for FSGS due to our patient’s long history of hypertension.
With unpublished data or lack of diagnostic investigation, it is likely that the number of cases of nephrotic syndrome associated with ADPKD is underreported. Conventionally, the presence of multiple bilateral renal cysts has been described as one of the relative contraindications to percutaneous renal biopsy due to difficulty assessing vascular anatomy; as such, many nephrologists and proceduralists may be reluctant to accept the risks of percutaneous renal biopsy in patients with ADPKD. In the literature, an open renal biopsy has been performed in most cases of ADPKD that warrant a biopsy, but many novel approaches have been described for use in “high-risk” scenarios where conventional contraindications to kidney biopsy may apply [6-8]. These include open, transvenous (transjugular or transfemoral), laparoscopic, or transurethral approaches [9]. Park and colleagues previously described a patient with ADPKD who was biopsied laparoscopically and suggested that the advantages of a laparoscopic approach include a sufficient diagnostic sample, minimal injury risk, hemostasis under direct vision, smaller incision, and a short hospital stay [4]. In our case, a discussion of risks and benefits with the patient as well as an experienced urologist at our center allowed the ultrasound-guided percutaneous biopsy to be done under general anesthesia in the operating room without complications.
Aside from the logistics of the biopsy procedure and interesting pathologic findings, this case also highlights the use of genetic testing to evaluate glomerular and cystic kidney disease. A monogenic cause of FSGS is relatively common and has implications for both therapy and transplant. In an adult cohort of patients with FSGS, a monogenic cause of disease was identified in 28% of patients with a family history of kidney disease and 8% of patients without a family history. Genetic testing can be used in patients with atypical presentations of polycystic kidney disease to help establish the diagnosis and has a role in living donor transplant evaluation. Our patient had a parent with a diagnosis of polycystic kidney disease and multiple bilateral cysts both of which are consistent with ADPKD; however, the small size of his kidney cysts and preserved kidney size are both atypical for ADPKD. We therefore proceeded with a genetic testing panel which evaluated for variants in genes known to cause both FSGS and cystic kidney disease. Consistent with our clinical diagnosis of secondary FSGS, testing was negative for variants in genes associated with FSGS. In addition, genetic testing for causes of cystic kidney disease was inconclusive. Our patient had a missense variant of unknown significance in PKD1—the gene mutated in the majority of patients with ADPKD. In addition, our patient had two identified variants in PKHD1—the gene mutated in Autosomal Recessive Polycystic Kidney Disease (ARPKD). Of the PKHD1 variants, one was a likely pathogenic protein-truncating variant and one was a missense variant of unknown significance in PKHD1. While ARPKD is often associated with severe complications in infancy or childhood; there is significant heterogeneity with many patients surviving into adulthood [10-12]. However, several factors argue against ARPKD in this case including the normal appearance of the liver on imaging and the father with polycystic kidneys which suggests an autosomal dominant mode of inheritance. No distal tubular cystic changes were seen. The lack of a definitive genetic diagnosis in individuals with suspected ADPKD is not unusual given substantial allelic heterogeneity. Truncating mutations in PKD1 and PKD2 are generally thought to be pathogenic, facilitating interpretation. In contrast, non-truncating variants including regulatory, missense, or splicing variants remain challenging to classify, as happened in the case of our patient.
In conclusion, we report a case of PKD with nephrotic-range proteinuria due to FSGS, which was diagnosed after percutaneous renal biopsy. When considering a biopsy to diagnose an additive renal process in PKD, it is essential to consider factors such as safety, adequacy, recovery time, and whether tissue diagnosis would impact management. Additionally, unusual clinical presentation or disease course in polycystic kidney disease may warrant genetic testing to differentiate between ADPKD and other causes of cystic kidney disease. Lastly, additional work is needed to help classify non-truncating variants in PKD1 and PKD2 to facilitate definitive genetic diagnosis and prognosis in ADPKD.
LV, GL, and MP were major contributors in writing the manuscript. GL provided expert input on the interpretation of genetic testing. VW performed a histological examination of the kidney and provided images that are included in the manuscript. All authors read and approved the final manuscript.
Declarations
Consent for publication: Consent for publication was obtained from the patient described in this case report.
Availability of data and materials: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Competing interests: MP reports consulting for Natera Inc., and Otsuka America Pharmaceutical Inc., and participating in Advisory boards for Otsuka America Pharmaceutical Inc. and Reata Pharmaceuticals Inc.
- Spithoven EM, Kramer A, Meijer E, Orskov B, Wanner C, Caskey F, Collart F, Finne P, Fogarty DG, Groothoff JW, Hoitsma A, Nogier MB, Postorino M, Ravani P, Zurriaga O, Jager KJ, Gansevoort RT; ERA-EDTA Registry; EuroCYST Consortium; WGIKD; EuroCYST Consortium; WGIKD. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. 2014 Dec;86(6):1244-52. doi: 10.1038/ki.2014.120. Epub 2014 May 14. PMID: 24827775.
- Chapman AB. Autosomal dominant polycystic kidney disease: time for a change? J Am Soc Nephrol. 2007 May;18(5):1399-407. doi: 10.1681/ASN.2007020155. Epub 2007 Apr 11. PMID: 17429048.
- Chapman AB, Johnson AM, Gabow PA, Schrier RW. Overt proteinuria and microalbuminuria in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1994 Dec;5(6):1349-54. doi: 10.1681/ASN.V561349. PMID: 7894001.
- Park JI, Lee H, An JN, Chin HJ, Kim S. Laparoscopic biopsy-proven lupus nephritis in autosomal dominant polycystic kidney disease. Kidney Res Clin Pract. 2012 Sep;31(3):192-5. doi: 10.1016/j.krcp.2012.06.002. Epub 2012 Jun 26. PMID: 26894026; PMCID: PMC4716091.
- Visciano B, Di Pietro RA, Rossano R, Mancini A, Zamboli P, Cianciaruso B, Pisani A. Nephrotic syndrome and autosomal dominant polycystic kidney disease. Clin Kidney J. 2012 Dec;5(6):508-11. doi: 10.1093/ckj/sfs147. Epub 2012 Nov 11. PMID: 26069794; PMCID: PMC4400568.
- Klahr S, Breyer JA, Beck GJ, Dennis VW, Hartman JA, Roth D, Steinman TI, Wang SR, Yamamoto ME. Dietary protein restriction, blood pressure control, and the progression of polycystic kidney disease. Modification of Diet in Renal Disease Study Group. J Am Soc Nephrol. 1995 Jun;5(12):2037-47. doi: 10.1681/ASN.V5122037. Erratum in: J Am Soc Nephrol 1995 Oct;6(4):1318. PMID: 7579052.
- Oda Y, Sawa N, Hasegawa E, Mizuno H, Kawada M, Sekine A, Hiramatsu R, Yamanouchi M, Hayami N, Suwabe T, Hoshino J, Takaichi K, Kinowaki K, Ohashi K, Fujii T, Ubara Y. PKD1-associated autosomal dominant polycystic kidney disease with glomerular cysts presenting with nephrotic syndrome caused by focal segmental glomerulosclerosis. BMC Nephrol. 2019 Aug 28;20(1):337. doi: 10.1186/s12882-019-1524-6. PMID: 31455242; PMCID: PMC6712641.
- Stiles KP, Yuan CM, Chung EM, Lyon RD, Lane JD, Abbott KC. Renal biopsy in high-risk patients with medical diseases of the kidney. Am J Kidney Dis. 2000 Aug;36(2):419-33. doi: 10.1053/ajkd.2000.8998. PMID: 10922324.
- Whittier WL, Korbet SM. Renal biopsy: update. Curr Opin Nephrol Hypertens. 2004 Nov;13(6):661-5. doi: 10.1097/00041552-200411000-00013. PMID: 15483458.
- Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, Consugar MB, Milliner DM, King BF, Torres VE, Harris PC. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006 Jan;85(1):1-21. doi: 10.1097/01.md.0000200165.90373.9a. PMID: 16523049.
- Gunay-Aygun M, Tuchman M, Font-Montgomery E, Lukose L, Edwards H, Garcia A, Ausavarat S, Ziegler SG, Piwnica-Worms K, Bryant J, Bernardini I, Fischer R, Huizing M, Guay-Woodford L, Gahl WA. PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol Genet Metab. 2010 Feb;99(2):160-73. doi: 10.1016/j.ymgme.2009.10.010. Epub 2009 Oct 20. PMID: 19914852; PMCID: PMC2818513.
- Burgmaier K, Kilian S, Bammens B, Benzing T, Billing H, Büscher A, Galiano M, Grundmann F, Klaus G, Mekahli D, Michel-Calemard L, Milosevski-Lomic G, Ranchin B, Sauerstein K, Schaefer S, Shroff R, Sterenborg R, Verbeeck S, Weber LT, Wicher D, Wühl E, Dötsch J, Schaefer F, Liebau MC. Clinical courses and complications of young adults with Autosomal Recessive Polycystic Kidney Disease (ARPKD). Sci Rep. 2019 May 28;9(1):7919. doi: 10.1038/s41598-019-43488-w. PMID: 31138820; PMCID: PMC6538621.