
More Information
Submitted: 20 November 2019 | Approved: 06 December 2019 | Published: 09 December 2019
How to cite this article: Gok F, Zeybek C, Balat A, Jones OY. A pilot study on treatment of infantile cystinosis with mesenchymal stem cells. J Clini Nephrol. 2019; 3: 181-185.
DOI: 10.29328/journal.jcn.1001046
Copyright License: © 2019 Gok F, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

A pilot study on treatment of infantile cystinosis with mesenchymal stem cells
Faysal Gok1*, Cengiz Zeybek2, Ayse Balat3 and Olcay Y Jones4
1Department of Pediatrics Pediatric Nephrology and Rheumatology, Medicana International Ankara, Hospital, Ankara, Turkey
2Department of Pediatric Nephrology, Gulhane University of Health Sciences, Etlik, Kecioren, 06100 Ankara, Turkey
3Department of Pediatric Nephrology, Faculty of Medicine, Istanbul Aydın University, Istanbul, Turkey
4Department of Pediatric Rheumatology, Walter Reed National Military Medical Center, Bethesda, MD, USA
*Address for Correspondence: Faysal Gok, Department of Pediatrics Pediatric Nephrology and Rheumatology, Medicana International Ankara, Ankara, Turkey, Tel: 90-312-292 92 92; 90-532 335 88 40; Fax: 90-312-287 20 01; Email: [email protected]
Infantile cystinosis is a lysosomal storage disease leading to end stage kidney disease at early ages. There is no effective treatment and patients require long term dialysis or kidney transplant for survival. We present our experience on three affected children who received HLA matched allogeneic stem cell transplant. The protocol used was novel and designed to promote engraftment. The primary endpoint was safety for treatment related mortality or morbidity; All three children survived without serious adverse effects during extended follow up for over 4 years. Although we could not prove engraftment, all three children met secondary end point of sustained target functions over a 6 month follow-up. Further studies are warranted to further evaluate safety and efficacy of MSC treatment for infantile cystinosis.
Cystinosis is a rare genetic disorder with autosomal recessive inheritance due to mutations of the CTNS gene on chromosome 17p13. It is a lysosomal storage disease and affects 1 to 2 out 100,000 live births a year. There are about 50 known lysosomal storage disorders (LSDs) and nearly 1/5th are caused by lysosomal membrane protein dysfunction [1]. Cystinosis is caused by a loss of function in cystinosin, a lysosomal membrane specific transport protein of cysteine. Cysteine is the oxidized dimer form of the amino acid cysteine produced after degradation of endocytosed protein. Increased concentrations of cysteine confined in the lysosomes lead to precipitations of cysteine crystals disrupting cellular oxidative metabolism and glutathione status, leading to altered mitochondrial energy metabolism, autophagy, and apoptosis overtime [2].
Cystinosin is ubiquitously expressed and most of the organs are affected as a result of metabolic changes. However, depending on the type of mutation in the CTNS gene, there are three clinical presentations of cystinosis [3,4]. Infantile cystinosis (OMIM 219800) constitutes the most common and severe form; The natural history of these children starts with failure to thrive, and polyuria within the first 4-6 months of life due to emerging renal tubular dysfunction (i.e. urinary loss of sodium, potassium, bicarbonate, magnesium, calcium, phosphate, glucose and low to intermediate molecular weight protein). Without treatment with cysteamine children develop renal insufficiency by 1 to 2 years of age when serum creatine is increased by 2 folds. Most children develop end stage renal disease and become dialysis dependent by the end of the first decade when serum creatinine > 10 mg/dL. In addition to renal findings, the patients also show corneal clouding and photophobia due to precipitations of cystine crystals by 2 years of age that can be followed by blindness due to retinal damage. In later stages these children also develop mutisystem failure including myopathy, neurologic and endocrine (including diabetes and thyroid) dysfunction, and cutaneous changes [5]. The two other forms of cystinosis follow a less severe course with slower progression compared to infantile form: juvenile cystinosis (OMIM 219900) is characterized by corneal and renal alterations after the 1st decade of life [6], and the ocular cystinosis (OMIM 219750) manifests with photophobia at adult ages [7].
The diagnosis of infantile cystinosis is based on clinical assessment for growth and slit lamp exam for corneal findings. The laboratory tests are limited to basic blood chemistries for kidney functions and urine analysis. Cysteine levels can be measured in peripheral blood leukocytes by tandem mass spectrometry, normal < 0.3 nmol/mg protein. There is no cure for cystinosis and current treatment is based on supportive measures and oral supplement of cysteamine (dose 1.30 to 1.95 g/m2/day to target leukocyte cysteine levels < 1.0 nmol/mg protein) and eye drops containing cysteamine. It is a chelator of cysteine that diffuses into lysosomes and facilitates cysteine transport. Cysteamine provides some, but limited, efficacy particularly during early phases of disease course, especially if started before the age of 5 years [8]. It delays disease progression, but is not a cure. Long term outcomes remain dominated by progress to organ failure and premature death in most cases. In the last few decades extension of life span was made possible by kidney transplant. However, concerns for paucity of donor kidney, long term immunosuppressive regimen, and financial cost bring additional concerns. Thus, there is an unmet need for development of new therapeutic modalities until gene treatment becomes a reality. For the time being, cell based treatment using healthy allogeneic donor stem cells is promising to treat infantile cystinosis. Although mesenchymal stem cells (MSC) have been tested in metabolic and degenerative diseases with some success [9,10], the literature on cystinosis remains limited to preclinical models. We now present our novel experience on treatment of three patients with infantile cystinosis using full matched adipose tissue derived MSC from healthy donors.
Patient selection and regulatory approvals
The patients were followed routinely by (Patient #1 and Patient #2) or referred from remote medical centers to (Patient #3) the pediatric nephrology clinic of Gulhane Medical Academy, Ankara Turkey. The study received proper regulatory approvals through the offices of Turkish Ministry of Health as well as institutional ethics council at Gulhane Medical Academy. Families were consulted in length on risk and benefits of the MSC transplant treatment prior to informed consent and informed assent on patients and MSC donors.
Donor MSC cell lines
HLA full match allogeneic adipose derived MSC was established separately for each patient. The selected healthy donors underwent a minor procedure of periumbilical subcutaneous fat tissue aspiration totaling about 15 ml volume. The procedure was done under local anesthesia and tolerated well. The primary MSC lines were prepared by LabCell
Treatment protocol
This is a pilot study on children (less than 18 years of age) with established diagnosis of cystinosis based on clinical presentation with vomiting, polyuria, polydipsia, exam findings of failure to thrive and corneal crytals, as well as laboratory evidence of proximal tubulopathy, and increased leukocyte cysteine levels. The primary end point objective was safety based on treatment-emergent adverse events using NCI common terminology criteria version 4.0; particular attention to serious adverse events. The secondary end point objective was sustained target organ functions at 6 month follow-up. The procedure was performed as under conscious sedation in operating room during overnight stay. Patients received MSC transplant (5 x 106 cells/kg per session) twice given at 3 week intervals. During each treatment, the cells were injected by 2 different routes: intra-arterial through catheterization (1 to 1.5 x 106/kg/dose) and intra bone marrow through the iliac bones (2-2.5 x 106/kg in right side and 2-2.5 x 106/kg in left side). Patient #1, received her intraarterial infusion through the intracoronary artery (first via the right coronary artery in session 1, then via the left coronary artery in session 2) to maximize myocardial exposure to MSC. Patient #2 and Patient #3 received intraarterial infusion through renal arteries bilaterally. The rational for the current procol design was to promote engraftment in bone marrow environment and accomplish long term donor chimerism. Furthermore, intra-arterial injection is known to maximize homing and tissue homeostasis. The follow-up included in person clinic visits, as well as, phone interviews for patient-parent reported outcomes.
Table 1 summarizes the information on three patients with established diagnosis of cystinosis. All three were term babies born to nonconsengious parents from different parts of Turkey. All three patients had similar presentation including normal weight and height at birth which progressed to failure to thrive during the first year of life. They presented to our clinic with complains of vomiting, polyuria and polydipsia. The laboratories showed abnormal urine analysis with evidence of proximal renal tubular dysfunction. Patient #3 was referred to our clinic at 6 months of age, soon after onset of poor weight gain. Family was fully aware of cystinosis as older sibling (3.5 year old brother) was also affected by the illness. On slit lamp exam, Patient #1 and Patient #2 had corneal crystals. All three patients had increased leukocyte cysteine levels at the time of diagnosis and were on cysteamine as a standard of treatment.
| Table 1: Cysteine levels measured in peripheral blood WBC. | |||
| Patient Properties | Patient #1 | Patient #2 | Patient #3 |
| Sex | F | M | F |
| Age at diagnosis | 8 months | 6 months | 6 months |
| CTNS gene mutations | Yes | Yes | Yes |
| Age at MSC treatment (0) | 8 years | 13.3 years | 8 months |
| Weight at Time 0 | 13 kg | 20 kg | 5 kg |
| Corneal crystals at Time 0 | Yes | Yes | No |
| Leukocyte cystein levels at 0 Time | 4.9 nmol/mg | 1.9 nmol/mg | 5.1 nmol/mg |
| Serum Cre at Time 0 | > 6 mg/dL | > 4.2 mg/dL | > 0.7 mg/dL |
| Site MSC Injections | Coronary artery & bone marrow | Renal artery & bone marrow | Renal artery & bone marrow |
| Total MSC Introduced per session | 1.5 x 106/kg | 1.5 x 106/kg | 1.5 x 106/kg |
| Number of total sessions (q 3 weeks) | 2 | 2 | 2 |
| Follow-up | 5 years | 4 years | 4 years |
| Concern leading to MSC | ESRD Cardiyomyopathy | chronic renal insuffciency | poor growth |
| Benfit of MSC | Stabilized homeostasis | Stabilized renal status | delayed renal prograss |
| Age at serum Cre > 1.4 | 4 years | 7 | N/A |
| Age at ESRD and start dialsis | 5.5 years | 14.3 | N/A |
| Age at renal transplant | 11 years | 15.3 | N/A |
Patient #1 had patchy compliance to cysteamine and progressed to developed chronic renal insufficiency around two years of age. By 5.5 years of age, she became anuric requiring peritoneal dialysis for ESRD. At 6 years of age she experienced dilated cardiomyopathy. Her clinical status progressed rapidly for signs of cardiac failure as her ejection fraction (EF) went down from 50% to 30% within months. While she was in renal transplant list, patient was recruited to receive mesenchymal stem cell treatment with a hope of stabilizing her homoestasis until availability of solid organ transplant. She received HLA full match (6/6) MSC derived from her 10 year old healthy brother and tolerated treatment well. The cardiac functions remained steady at EF of 30% without further detoriation until she underwent renal transplant at 11 years of age. Figure 1 shows her leukocyte cysteine levels nadiring at 5 months post MSC followed by gradual uptrend. Patient #2 was compliant with cysteamine as evidenced by slow progression of renal insufficiency. He received full matched MSC from 15 years old healthy sister that improved his overall stigma and quality of life by patient and physician global assessment. His leukocyte cysteine levels decreased from 1.9 to 0.44 nmol/mg protein and renal functions remained stable (serum creatine at 4.21 mg/dL) during the first 6 months post MSC. However, he did progressed to ESRD by 12 months after treatment suggesting MSC was not effective for tissue remodeling. Later he received renal transplant and currently doing well. Based on encouraging experience on Patient #1 and Patient #2 for safety, Patient #3 received MSC during infancy before the onset of irresible tissue damage with a hope to prevent disease progression. She was stable for the first 6 months post-transplant but also progressed over time. At the end of 4 year follow-up, her growth parameters are < 3% for age (height 81 cm and weight 11 kg), serum creatine is 0,86 mg/dL and recent eye exam is positive for cysteine crystals. Nonetheless, her course has been slower compared to her brother who had serum creatine of 3.2 mg/dL at 4 years of age. The parents are forthcoming with similar conclusions and inquiring if repeat MSC treatment is available.
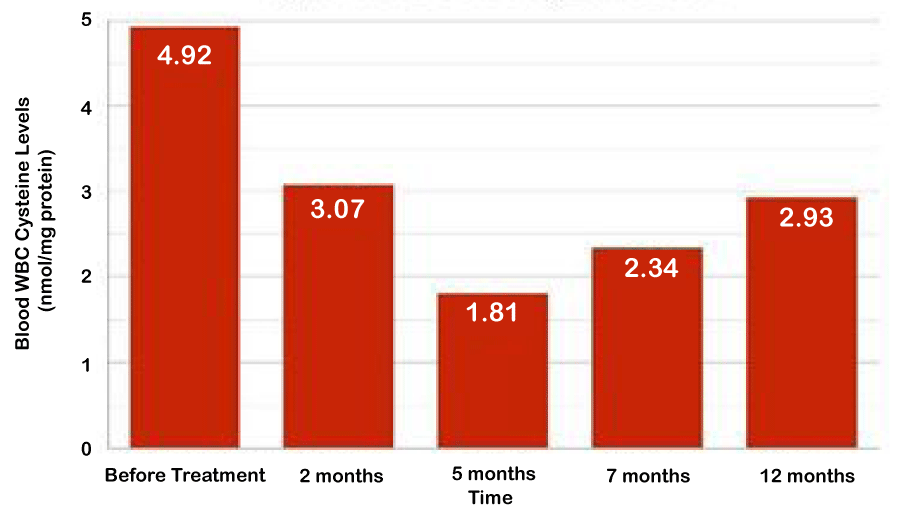
Figure 1: Blood WBC Cysteine Levels.
In summary, all three patients tolerated the MSC treatments well. There was no evidence of treatment related complications including thromboembolic events, myocardial infarct, pulmonary emboli, stroke, bone marrow concerns, infections, ectopic growth or malignancy. In addition, MSC treatment provided supportive measures: in older hosts with pending organ failure, it provided sustained homeostatic stability (Patient #1 & Patient #2) and in a young host with normal kidney function (Patient #3), it slowed the progression of organ damage.
To our knowledge, this is the first clinical study on treatment of infantile cystinosis with MSC transplant. The procedure was tolerated well for both patients and donor siblings. The treatment was safe for all three children even when it was given in the presence of organ compromise or during the first year of life. There was no adverse effect in short (24 hours) or long term (up to 5 years) follow-up. Furthermore, secondary endpoint of progress free survival for 6 months post treatment was also accomplished in all three patients. The protocol that we developed was novel and based on delivery of cells both intra-arterially and intra-bone marrow injections that resulted with decreased leukocyte cysteine levels. Although Patient #1 remained in ESRD, the gained window time allowed long enough survival until availability of donor kidney for solid organ transplant. Similarly, Patient #2 who was in the brim of ESRD was able to prolong his status for a year prior to ESRD. This contributed in shortened duration of dialysis dependence prior to availability of donor kidney for renal transplant. Sustained homeostasis was most prominent in Patient #3 whose renal compromise delayed significantly in long term follow-up compared to the course of her older brother with infantile cystinosis.
We believe the targeted delivery directly into the arterial flow as well as into the bone marrow was beneficial to maximize homing of MSC. Decreased cysteine levels in blood leukocytes is likely to be based on interactions between the donor stromal and hematopoietic cells in the bone marrow sinusoidal. These progeny cells can facilitate the delivery of cargo to periphery upon emigration from the bone marrow. Our findings are compatible with the preclinical studies on cystinosis models; In the study by Syres, et al., C57BL/6 CTNS-/- mice treated with single intravenous injection of non-fractionated bone marrow cells (BMC; 2 x 107 per mouse) or ex vivo expanded bone marrow derived MSC (1 x 106 per mouse) [11]. These mice received conditioning regimen with lethal (cesium radiation, 8 Gy) or non-lethal (3.6 Gy) total body radiation prior to transplant of BMC or MSC, respectively. The donor cells were derived from healthy green florescent (GFP) transgenic C57BL/6 mice. Post mortem examination of tissues showed significant engraftment of GFP positive cells (up to 13%) throughout tissues in mice treated with BMC but not those treated with MSC (< 5%). Regardless of the percent engraftment, at 4 month follow-up, the levels of cysteine in brain, heart, and kidneys were significantly lower both in BMC (down by 56%, 82% and 70%, respectively) and MSC (down by 56%, 59% and 49%, respectively) treated mice compared to untreated controls. Similarly, in our studies, we could not find evidence of donor chimerism in Patient #1 or in Patient #2 (using Y or XX chromosome markers, respectively) in bone marrow aspirates at Time 6 months (data not shown). Nonetheless, the clinical course of Patient #3 suggests possible engraftment that may be below the detectable levels. Although it is not accustom to use conditioning before MSC treatment in clinical trials, crafting protocols further may enhance engraftment. Furthermore, the effects of MSC can be attributed to paracrine effects: in an in vitro study [12], the levels of cysteine in cells derived from patients with cystinosis was decreased when co-cultured with MSC. This was shown to be mediated by micro-vesicles measuring 100-400 nm in diameter released from MSC that carry cystinosin protein and mRNA as cargo. This is encouraging to develop cell free products of MSC that may further increase safety of cell treatment particularly if protocols require long term repeat injections starting from infancy.
In summary, the results of this pilot study suggest MSC treatment of cystinosis is safe and provide periods of stability at varying extends that correlates inversely with existing tissue damage. Although, MSC could not prevent progression of cystinosis, it can slow the histopathologic changes, albeit at limited extend, particularly if it is given at young ages. This is probably by paracrine mechanisms of MSC and further investigations are warranted to harness this property by optimizing and protocols and stem cell derived products.
Disclaimer
The views expressed in this article are those of the author and do not reflect the official policy of the Department of Army/Navy/Air Force, Department of Defense, or U.S. Government.
We extend our sincere appreciations to Dr. Ercument Ovali, Director, LabCell, Acibadem University, Dr. Ayhan Kilic, Director, Pediatric Cardiology, Gulhane University for their invaluable help in the study and to William A. Gahl, NIH/NHGRI for his critical review of the manuscript.
- Ruivo R. Anne C, Sagne C, Gasnier B. Molecular and cellular basis of lysosomal transmembrane protein dysfunction. Biochem Biophys Acta. 2009; 1793: 636–649. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19146888
- Sansanwal P, Sarwal MM. Abnormal mitochondrial autophagy in nephropathic cystinosis. Autophagy. 2010; 6: 971-973. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20729635
- Anikster Y, Shotelersuk V, Gahl WA. CTNS mutations in patients with cystinosis. Hum Mutat. 1999; 14: 454–458. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10571941
- Attard M, Jean G, Forestier L, Cherqui S, van't Hoff W, et al. Severity of phenotype in cystinosis varies with mutations in the CTNS gene: predicted effect on the model of cystinosin. Hum Mol Genet. 1999; 8: 2507–2514. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10556299
- Gahl WA, Thoene JG, Schneider JA. Cystinosis. New Engl J Med. 2002; 347: 111–121. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12110740
- Goldman H, Scriver CR, Aaron K, Delvin E, Canlas Z. Adolescent cystinosis: comparisons with infantile and adult forms. Pediatrics. 1971; 47: 979–988. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/5141767
- Cogan DG, Kuwabara T, Kinoshita J, Sheehan L, Merola L. Cystinosis in an adult. J Am Med Assoc. 1957; 164: 394–396. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/13415995
- Brodin-Sartorius A, Tete MJ, Niaudet P, Antignac C, Guest G, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012; 81: 179–189. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21900880
- Rocca CJ, Cherqui S. Potential use of stem cells as a therapy for cystinosis. Pediatr Nephrol. 2019; 34: 965-973. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29789935
- Cahill RA, Jones OY, Klemperer M, Steele A, Mueller TO, et al. Replacement of recipient stromal/mesenchymal cells after bone marrow transplantation using bone fragments and cultured osteoblast-like cells. Biol Blood Marrow Transplant. 2004; 10: 709-717. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15389437
- Syres K, Harrison F, Tadlock M, Jester JV, Simpson J, et al. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood. 2009; 114: 2542–2552. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19506297
- Iglesias DM, El-Kares R, Taranta A, Bellomo F, Emma F, et al. Stem Cell Microvesicles Transfer Cystinosin to Human Cystinotic Cells and Reduce Cy stine Accumulation In vitro. PLoS One. 2012; 7: e42840. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22912749