
More Information
Submitted: 01 November 2019 | Approved: 19 November 2019 | Published: 20 November 2019
How to cite this article: Fonseca JA, Jorge S, Gameiro J, Lopes JA, Guerra JA. Diabetes and chronic kidney disease: a rare cause for a very common association. J Clini Nephrol. 2019; 3: 164-167.
DOI: 10.29328/journal.jcn.1001043
Copyright License: © 2019 Fonseca JA, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Diabetes and chronic kidney disease: a rare cause for a very common association
José Agapito Fonseca*, Sofia Jorge, Joana Gameiro, José António Lopes and José Oliveira Guerra
Department of Nephrology and Renal Transplant, Av. Professor Egas Moniz, Hospital de Santa Maria, 1649-035, Lisbon, Portugal
*Address for Correspondence: José Agapito Fonseca, Rua General Pimenta de Castro, nº13, 6ºEsq 1700-217 Lisboa, Portugal, Tel: +351913137104; Email: [email protected]
The authors present the case of a 45-year-old female patient with diabetes and chronic kidney disease (CKD). She had unsatisfactory glycemic control, and showed some intellectual limitations. Her urine exam was unremarkable, and her renal ultrasound revealed single right kidney with aspects suggesting ureteropelvic junction syndrome. Her mother had also suffered from diabetes and CKD G5D presenting in the sixth to seventh decade.
An hereditary cause for CKD was considered, which led the authors to investigate an autosomal dominant cause for CKD with a tubulointerstitial phenotype, taking into account the personal and family history for diabetes and also the renal imaging; a large deletion in the HNF-1β gene was identified through Multiplex Ligand Probe Assay (MLPA) analysis, explaining the phenotype.
Genetic causes of CKD should be considered in the presence of positive family history for CKD, and the coexistence of diabetes with bland urine sediment should raise the possibility of a syndromic cause of the phenotype, namely involving HNF-1β gene mutations or deletions.
Inherited kidney diseases are frequently undiagnosed, but they affect nearly 10% of adults and the majority of children who require renal replacement therapy [1]. Recent advances in molecular and genetic testing have allowed for an increase in the establishment of the correct genetic diagnosis of some hereditary chronic kidney diseases, leading to an improvement in medical care and opening new perspectives, namely therapeutic, in an expected near future. In fact, a correct genetic diagnosis may represent a positive step in the individual medical approach, concerning prognosis, reproductive planning and counseling for relatives at risk. However, genotype-phenotype correlations are not always clear, turning clinical practice into an increasing challenge.
For nephrologists, diabetes is a well-known cause of chronic kidney disease (CKD), but not all diabetic patients suffer from “classical” diabetic nephropathy. Hereditary diabetes and kidney disease should be considered in the presence of positive family history, and a bland urine sediment with slight or no proteinuria points to tubulointerstitial phenotype; in the investigation of hereditary diabetes and CKD, the mutations/deletions in HNF-1β gene, as well as mitochondrial deafness, particularly maternally inherited diabetes and deafness (MIDD), should be considered. The renal phenotype, including urine sediment and imaging aspects, can give important clues for the correct diagnosis.
A 45-year-old caucasian female was admitted to our emergency department for syncope. She had past medical history of diabetes mellitus diagnosed 14 years before, under insulin treatment over the past 2 years with unsatisfactory metabolic control, without known target-organ disease, and hypothyroidism. Besides insulin, linagliptin and levothyroxine, she did not take other medications. She denied previous use of unprescribed medication, namely NSAIDs, or herbal medications; she denied foamy or hematic urine, polyuria/polydipsia previous to the diabetes diagnosis, or recurrent respiratory or urinary tract infections, cutaneous or articular complaints (oral ulcers, alopecia, rash, arthralgias, myalgias), red eye/other ocular complaints; her audition showed no compromise, she denied reduced visual accuracy; she also denied neurologic symptoms, or past medical history of epilepsy or stroke-like episodes.
Her 82-year-old mother was also diabetic, suffered from chronic kidney disease, and had recently started on dialysis. Her 43-year-old brother was also diabetic. She had three healthy children.
The patient´s physical exam was unremarkable, except for slight conjunctival pallor and a relatively poor speech, suggestive of possible cognitive compromise, unrecognized by her or her family (she was unemployed and had basic scholarship, married). Fundoscopy was negative for diabetic retinopathy.
The laboratorial evaluation revealed urea 114 mg/dL, serum creatinine 4,5 mg/dL; potassium (4,5 mmol/L), magnesium (1,7 mmol/L), uric acid (4,9 mg/dL) were in the normal range, as well as liver function tests; her urine exam revealed hemato-proteinuria, with 24h proteinuria of 187 mg; renal ultrasound revealed enlarged right kidney, measuring approximately 170 mm with pelvicalyceal dilation, reduced renal parenchyma and slightly increased echogenicity, suggesting ureteropelvic junction syndrome (Figure 1); the left kidney was not visualized, neither the left ureter.
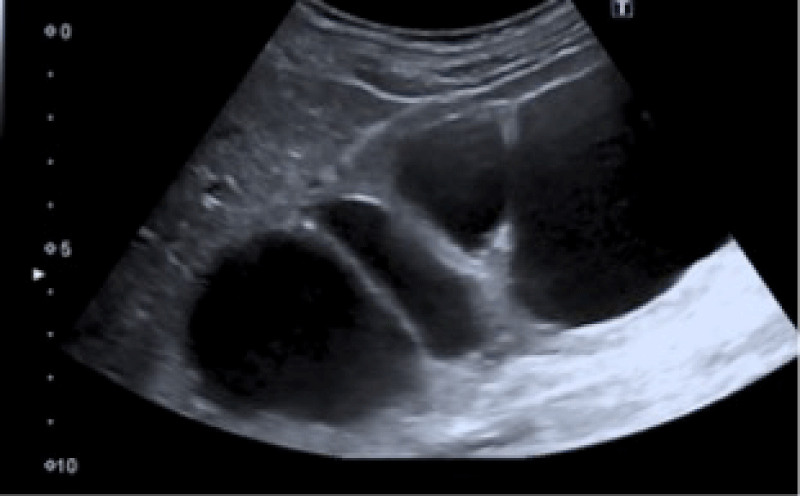
Figure 1: The left kidney was not visualized, neither the left ureter.
Previous laboratorial examination was obtained, revealing serum creatinine of 2 mg/dL one year before.
Considering chronic kidney disease of unknown etiology, complimentary investigation was performed: viral serologies for HIV 1 and 2, HBV and HCV were negative; immunologic tests were negative (ANA, antids DNA, cANCA and pANCA, lupic anticoagulant, anticardiolipin antibodies, rheumatoid factor, cryoglobulins), complement C3 and C4 levels were in the normal range, ASO titer was normal; serum protein electrophoresis and serum free light chain levels were unremarkable.
A renogram and renal scintigraphy were performed, revealing moderate to severe renal impairment of the right kidney, and the renogram with diuretic challenge was compatible with ureteropelvic junction syndrome.
Considering these data, as well as the family history, the hypothesis of hereditary CKD was considered, namely autosomal dominant tubulointerstitial kidney disease (ADTKD), specifically related to HNF-1β gene, or mitochondrial disease, namely MIDD.
Genetic testing was performed, with negative results for MIDD, but a large deletion of the hepatocyte nuclear factor-1β (HNF-1β) gene was identified through the use of MLPA (kit P241-D2, MRC-Holland).
This result is consistent with the phenotype described, leading to the diagnosis of ADTKD due to a large deletion of the HNF-1β gene, with extra-renal manifestations corresponding to a syndromic form of TGF-β associated disease.
Due to severe kidney impairment and stable function, the Urology department did not consider pyeloplastic surgery or other urinary diversion surgery. The patient maintained regular follow up at the Nephrology consultation with an observation strategy, but kidney function deteriorated during the three years of follow up, with the patient starting hemodialysis.
The presentation of CKD with a bland urine exam, with slight hematuria and proteinuria, is consistent with tubulointerstitial phenotype [2]. The ureteropelvic junction syndrome suggests possible congenital abnormality of the genito-urinary tract (CAKUT). The family history of diabetes and CKD were consistent with an autosomal dominant or maternal inheritance pattern, possibly syndromic: bland urine sediment, with absent microalbuminuria/proteinuria, absence of retinopathy, and urinary tract abnormalities.
Considering the tubulointerstitial phenotype and inheritance pattern, mitochondrial diseases such as MIDD could be considered; in MIDD kidney disease can occur with tubular or glomerular involvement, with simultaneous affection of other organs, causing deafness, myocardial hypertrophy and epilepsy or stroke-like episodes; due to heteroplasmy, the clinical manifestations can be quite different in different family members, which could justify the absence of deafness or epilepsy or stroke like episodes, in the presence of a renal tubulointerstitial phenotype and diabetes in our patient. However, MIDD would not explain the ureteropelvic junction syndrome.
Also implicated in hereditary AD CKD with tubulointerstitial phenotype are ADTKD, namely involving HNF-1β gene. The HNF-1β gene mutations have been described as a possible cause of syndromic CKD or autosomal dominant TIN only with renal disease2. Extra-renal phenotypes include early-onset diabetes mellitus, pancreatic hypoplasia, urogenital tract malformations, abnormal liver function, early-onset gout, central nervous system manifestations or even autism spectrum disturbances, probably due to the fact that HNF-1β is a transcription factor that regulates multiple genes expressed in several organs, including kidney, pancreas, and liver [3-5]. Mutations in this gene represent the most common known monogenic cause of developmental kidney disease [6,7], implicated in about 10% of the cases of congenital abnormalities of the kidney and urinary tract (CAKUT) [6]. Heterozygous mutations in the coding region or splice sites of HNF-1β, and complete gene deletion, each account for approximately 50% of all cases of HNF-1β-associated disease [6-9]. The severity of the renal disease associated with HNF-1β mutations is extremely variable, from prenatal renal failure to normal renal function in adulthood, and there is no clear genotype-phenotype correlation [6-11], consistent with haploinsufficiency as the disease mechanism. Renal manifestations include a spectrum of possible morphological abnormalities such as multicystic, dysplastic, hypoplastic kidneys, renal agenesis, ectopic kidney, reflux, hydronephrosis, and also prune belly syndrome; electrolyte abnormalities, such as hypomagnesemia and hypokalemia can also occur [6.7], and chronic kidney disease due to interstitial fibrosis, with a highly variable rate of decline of estimated glomerular filtration rate. Hyperuricemia can be a precocious manifestation, as well as hypomagnesemia. In fact, although 50%-60% of ADTKD- HNF-1β patients develop hypomagnesemia, HNF-1β mutations are mainly identified in patients with structural kidney defects or diabetes [12-14]. Interestingly, HNF1B-related nephropathy is proposed to be a mitochondrial disorder in adulthood [13].
Our patient presented with a neurobehavioural disorder, which can also be explained by the HNF-1β deletion; an increase in neurodevelopmental disorders has been observed in individuals harbouring the 17q12 deletion but not in patients with HNF-1β coding mutations [15].
Data from animal models suggest that HNF-1β has an important function during several stages of nephrogenesis; however, the precise signaling pathways remain to be elucidated. In the embryonic kidney, HNF-1β is required for ureteric bud branching, initiation of nephrogenesis, and nephron segmentation. Ablation of mouse HNF-1β in nephron progenitors causes defective tubulogenesis, whereas later inactivation in elongating tubules leads to cyst formation due to downregulation of cystic disease genes, including Umod, Pkhd1, and Pkd2. In the adult kidney, HNF-1β controls the expression of genes required for intrarenal metabolism and solute transport by tubular epithelial cells. Tubular abnormalities observed in HNF-1β nephropathy include hyperuricemia with or without gout, hypokalemia, hypomagnesemia, and polyuria. Mechanism of fibrosis has also been recently explained, through ablation of HNF-1β in renal epithelial cells, leading to activation of a Twist 2-dependent transcriptional network that induces epithelial-mesenchymal transition and aberrant TGF-β signaling, resulting in renal fibrosis. Recent studies have identified novel post-transcriptional and post-translational regulatory mechanisms that control HNF-1β expression and activity, including the miRNA cluster miR17 ∼92 and the interacting proteins PCBD1 and zyxin. Further understanding of the molecular mechanisms upstream and downstream of HNF-1β may lead to the development of new therapeutic approaches in cystic kidney disease and other HNF1B-related renal disease [15-17].
HNF-1β coordinates transcriptional networks involved in nephrogenesis, epithelial differentiation, tubular transport, and intrarenal metabolism. Therefore, patients who carry heterozygous mutations in TGF-β may present with complex renal phenotypes. To date, genotype-phenotype studies have not found a correlation between the type of mutations and the type and/or severity of renal disease, and patients who harbor the same TGF-β mutation show inter- and intrafamilial variability [12].
For the nephrologists, facing the association of diabetes and CKD is a common challenge, with most of the cases of renal affection being due to diabetic nephropathy or the coexistence of hypertension or ischemic kidney disease. However, the nephrologist must be aware of these rarer forms of diabetes and kidney disease, particularly when the urine sediment is bland, nor compatible with the classical diabetic nephropathy with predominant glomerular affection and consequent albuminuria progressing to massive proteinuria.
The presence of multiorganic involvement, in the presence of diabetes and CKD with hereditary involvement, should also be a clue for the nephrologist to suspect for a genetic syndromic diagnosis for CKD.
Given the costs of a genetic study, in recent years Faguer, et al., have proposed a score to predict the likelihood of having HNF1 beta gene compromise [12], but it does not include neuropsychiatric manifestations, and we presently are aware of the possible single renal phenotype [1]. Given the lower cost and availability of new genetic testing, we believe that an individual approach should be given to each patient.
In conclusion, the authors would like to emphasize the importance of considering genetic causes of CKD in face of a positive family history, as well as in the presence of syndromic phenotypes. Genetic screening tests are a promising and powerful tool in identifying the cause of chronic kidney disease, particularly of those with unknown etiology [18]. HNF-1β molecular defects in particular, should be considered when diabetes (MODY) and chronic kidney disease are present, with a tubulointerstitial presentation, and an autosomal dominant inheritance pattern [1].
Compliance with ethical standards
On behalf of all authors, the corresponding author states that there is no conflict of interest.
- Mehta L, Jim B. Hereditary Renal Diseases. Semin Nephrol. 2017; 37: 354-361. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28711074
- Bleyer A, Kmoch S. Autosomal dominant tubulointerstitial kidney disease: of names and genes. Kidney Int. 2014; 86: 459–461. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25168494
- Eckardt KU, Alper SL, Antignac C, Bleyer AJ, Chauveau D, et al. Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management—A KDIGO consensus report. Kidney Int. 2015; 88: 676-683. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25738250
- Coffinier C, Barra J, Babinet C, Yaniv M. Expression of the vHNF1/HNF1beta homeoprotein gene during mouse organogenesis. Mech Dev. 1999; 89: 211–213. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10559500
- Kolatsi-Joannou M, Bingham C, Ellard S, Bulman MP, Allen LI, et al. Hepatocyte nuclear factor-1beta: a new kindred with renal cysts and diabetes and gene expression in normal human development. J Am Soc Nephrol. 2001; 12: 2175–2180. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11562418
- Desgrange A, Heliot C, Skovorodkin I, Akram SU, Heikkilä J, et al. HNF1B controls epithelial organization and cell polarity during ureteric bud branching and collecting duct morphogenesis. Development. 2017; 15: 144: 4704-4719. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29158444
- Raaijmakers A, Corveleyn A, Devriendt K, van Tienoven TP, Allegaert K, et al. Criteria for HNF1B analysis in patients with congenital abnormalities of kidney and urinary tract. Nephrol Dial Transplant. 2015; 30: 835-842. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25500806
- Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat Rev Nephrol. 2015; 11: 102-112. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25536396
- Bockenhauer D, Jaureguiberry G . HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol. 2016; 31: 707-714. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26160100
- Heidet L, Decramer S, Pawtowski A, Morinière V, Bandin F, et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol. 2010; 5: 1079-1090. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20378641
- Weber S, Moriniere V, Knuppel T, Charbit M, Dusek J, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006; 17: 2864–2870. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16971658
- Ulinski T, Bensman A, Lescure S. Abnormalities of hepatocyte nuclear factor (HNF)-1beta: biological mechanisms, phenotypes, and clinical consequences. Arch Pediatr. 2009; 16: 1049-1056. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19361964
- van der Made CI, Hoorn EJ, de la Faille R, Karaaslan H, Knoers NV, et al. Hypomagnesemia as First Clinical Manifestation of ADTKD-HNF1B: A Case Series and Literature Review. Am J Nephrol. 2015; 42: 85-90. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26340261
- Decramer S, Parant O, Beaufils S, Clauin S, Guillou C, et al. Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J Am Soc Nephrol. 2007; 18: 923–993. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17267738
- Clissold RL, Ashfield B, Burrage J, Hannon E, Bingham C, et al. Genome-wide methylomic analysis in individuals with HNF1B intragenic mutation and 17q12 microdeletion. Clin Epigenetics. 2018; 10: 97. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30021660
- Faguer S, Chassaing N, Bandin F, Prouheze C, Garnier A, et al. The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int. 2014; 86: 1007-1015. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24897035
- Chan SC, Zhang Y, Shao A, Avdulov S, Herrera J, et al. Mechanism of fibrosis in HNF1 Brelated autosomal dominant tubulointerstitial kidney disease. J Am Soc Nephrol. 2018; 29: 2493–2509. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30097458
- Lata S, Marasa M, Li Y, Fasel DA, Groopman E, et al. Whole-exome sequencing in adults with chronic kidney disease: a pilot study. Ann Intern Med. 2018; 168; 100–109. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29204651