Case Report
Acute Tubulointerstitial Nephritis due to Phenytoin: Case Report
Nilzete Liberato Bresolin1*, Pedro Docusse Junior2, Maria Beatriz Cacese Shiozawa3, Marina Ratier de Brito Moreira4 and Natalia Galbiatti Silveira5
1Pediatric Nephrologist and Critical Care at de Gusmão Children’s Hospital. Professor at Federal University of Santa Catarina, ALANEPE Council, Brazil
2Medicine Student at Federal University of Santa Catarina, Brazil
3Maria Beatriz Cacese Shiozawa - Pathologist and Professor at Federal University of Santa Catarina, Brazil
4Marina Ratier de Brito Moreira - Pediatric Critical Care at Joana de Gusmão Children’s Hospital, Brazil
5Natalia Galbiatti Silveira - Pediatrician at Joana de Gusmão Children’s Hospital, Brazil
*Address for Correspondence: Nilzete Liberato Bresolin, Pediatric Nephrologist and Critical Care at de Gusmão Children’s Hospital, Professor at Federal University of Santa Catarina, ALANEPE Council, Brazil, Email: [email protected]
Dates: Submitted: 04 March 2017; Approved: 12 June 2017; Published: 13 June 2017
How to cite this article: Bresolin NL, Docusse P. Jr, Shiozawa MBC, de Brito Moreira MR, Silveira NG. Acute Tubulointerstitial Nephritis due to Phenytoin: Case Report. J Clini Nephrol. 2017; 1: 019-025. DOI: 10.29328/journal.jcn.1001004
Copyright License: © 2017 Bresolin NL, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Acute kidney injury; Paediatrics; Multiple trauma; Interstitial nephritis; Phenytoin
SUMMARY
Introduction: Acute tubulointerstitial nephritis (ATIN) is an acute kidney injury (AKI) resulting from damage to the tubulointerstitial tissue due to infection, trauma, or use of medication. It is clinically non-specific.
Case: A teenager with multiple trauma, hospitalised after lowering of level of conscience, and convulsion fits. While in the emergency ward, he received: midazolam, fentanyl and phenytoin. The cranial and abdominal CT scans were normal. He was stable with no signs of shock, trauma or infection; he developed oliguria and serum creatinine (Scr) 1.7mg/dL), 12 hours after the admission. After 36 hours, Scr levels were at 3.4mg/dL and urea at 55mg/dL. He had AKI according to pRIFLE (66.2% reduction in clearance). After other causes of AKI had been ruled out, the possibility of ATIN was raised; the phenytoin was suspended and pulse therapy, with methylprednisolone, was promptly initiated. After the first pulse, there was already a decline in the creatinine and urea readings; 48 hours later: Scr at 2.2mg/dL and urea at 86mg/dL. Thirty days after being discharged from hospital, the patient was in good health and had full restoration of kidney function.
Discussion: The singularity of this report relies on the rarity of ATIN secondary to the use of phenytoin and also in the importance of recognizing this aetiology as being one of the origins of AKI.
Conclusion: Early diagnosis allows the reversal of AKI through suppression of treatment with phenytoin and introduction of corticosteroid therapy, when necessary.
INTRODUCTION
Acute kidney injury (AKI) is characterised by the sudden and normally reversible reduction of kidney function, with loss of the capacity for the organism’s homoeostasis to be maintained. This can be accompanied, or not, by a reduction in diuresis [1]. The occurrence of AKI is very high among patients who are seriously ill [2]. At present, the criteria known by the acronym RIFLE (Risk, Injury, Failure, Loss, End Stage Renal Disease) are widely used to establish the diagnosis and the classification of AKI. There is a modified version of this criterion, specially used for paediatric patients, which has become known as pRIFLE (Paediatric Risk, Injury, Failure, Loss, End Stage Renal Disease), based on the reduction of creatinine clearance (ClCr) as estimated by Schwartz’s formula and/or a reduction of the urinary output based on body weight per hour (whichever of the criteria gives the worse result) [1].
In paediatrics, the main causes of AKI are infections, use of medication, and haemodynamic malfunction. In patients who have suffered multiple trauma, AKI can be caused by a multiplicity of factors, involving: poor volemia, rhabdomyolysis, abdominal compartment syndrome, sepsis; exposure to radiological contrast and to potentially nephrotoxic drugs, particularly antibiotics and antifungal agents [3].
Acute tubulointerstitial nephritis (ATIN) is an important cause of AKI, characterised, from the anatomopathological standpoint, by an interstitial infiltrate of mononuclear cells and eosinophils [4,5]. The classical clinical presentation of cases of ATIN includes the following key signs and symptoms: fever, skin rash, eosinophilia, and non-oliguric kidney failure, together with tubular dysfunction and abnormalities in the urinary sediment [4]. However, most cases proceed with non-specific symptoms, only followed by insidious deterioration of kidney function [6-8]. Probable causes include therapy based on medication; infection; and self-immune diseases [8]. Currently, most of the cases (about 70% of all cases) are related, in some way, to therapy based on medication [9]. There is a wide variety of drugs and medicines that could potentially cause ATIN [10,11].
The aim of the present study is to describe the case of a paediatric patient, who has suffered multiple trauma, and who developed AKI through ATIN secondary to the use of phenytoin, a drug used to treat convulsion and epilepsy, derived from the hydantoin class of anticonvulsant drugs [12]. So far, 5 cases of association between phenytoin and ATIN have been recorded in specialised literature throughout the world [12-16 ].
REPORT
A 14-year-old boy, of the Black race, weighing 60 kg and 1.64 metres tall, was admitted to the emergency unit at the Joana de Gusmão Children’s Hospital after having been run over. At the site of the accident, he showed lowering of the level of consciousness and also mental confusion. During his transfer to the hospital, this developed further, with two generalised tonico-clonic convulsive fits, each lasting 50 seconds, and showing a score of 7 points on the Glasgow Coma Scale (GCS). At the emergency unit, he was given medication, in preparation for a rapid sequence of intubation: midazolam (4x 0.1mg/kg/dose) and fentanyl (1x 1mcg/kg/dose) and then, as treatment and prophylaxis against convulsions, there was the start of treatment with phenytoin applied intravenously (attack dose of 20mg/kg) and maintenance (5mg/kg/day). The CT scans on the brain, the spine and the abdomen, as performed on admittance to the hospital, showed normal results. The patient was haemodynamically stable and afebrile, meaning no need for vasoactive or antibiotic drugs; he was given a urinary catheter and then sent on to the Intensive Care Unit (ICU) to be monitored. The results of his Complete Blood Count (CBC) test included: Red blood cell (RBC) count normal (haemoglobin at 13.4g/dL; haematocrit at 39%); White blood cell (WBC) count showed an increase in leukocytes (23940/mm3) as follows: rods 6%; segmented 80%; Platelets at 272000; Acidotic arterial gasometry (pH of 7.26; pCO2 29mmHg; pO2 57mmHg; HCO3 13mmol/L; Beb -12.5mmol/L); Partial urine test with macroscopic haematuria (red blood cells >10⁶/mL and Hb +); normal coagulation; Lactic acid test at 21mg/dL [RR 5.5-22mg/dL]; Creatine phosphokinase test at 102mcg/L [RR 10-120mcg/L]; Serum creatinine (Scr) at 1.1mg/dL, with the patient also showing a basal creatinine clearance (ClCr) of 104.3 mL/min/1.73m²SC, urea at 54mg/dL; electrolytes, kidney function, amylase and lipase, all normal.
On the first day of hospitalisation in the intensive care unit (ICU), 12 hours after being admitted to hospital, the patient had diuresis at 1ml/kg/hour and Scr at 1.7mg/dL, which increased steadily after that, with Scr at 2.5mg/dL six hours later. The new laboratory results showed: Haemoglobin at 13.5g/dL; Haematocrit at 38.5%; Leukocytes at 10870mm³ (rods 1%; segmented 80%); Platelets at 153000; Normal arterial gasometry (pH of 7.47; pCO2 45mmHg; pO2 63.2 mmHg; HCO3 32.9 mmol/L) and; Partial urine test just with microscopic haematuria. After 36 hours, the control Scr was at 3.4mg/dL and urea at 55mg/dL; according to the classification proposed by the pRIFLE criteria, the patient already had kidney injury with the loss of 66.2% of renal function and ClCr of 33.7mL/min/1.73m2SC. Forty-eight hours after hospitalisation, the patient kept his diuresis but still showed increased urea and Scr (64mg/dL and 4.2mg/dL respectively). An ultrasound scan on the urinary tract was then performed, showing no abnormalities.
Due to the unfolding of the case and ruling out other causes for acute kidney injury (AKI) as: crush syndrome, bleeding, septic and hypovolemic shock, infections and the using of commons drugs, and with phenytoin being the only medication received which could be connected, in some way, a diagnosis of acute tubulointerstitial nephritis was considered. In the light of this, a renal biopsy was requested. However, as it was impossible to conduct the biopsy at that moment, and also due to the progressive malfunction caused by the kidney injury, it was decided to stop the application of phenytoin; in addition, there being no improvement to kidney function, it was decided to start pulse therapy with methylprednisolone (1 gram per day). Other medical tests were also requested, namely: serum dose of total complement and fractions. HIV tests; tests for Hepatitis B and C; toxoplasmosis; cytomegalovirus; FAN; and anti-DNA. The results of all these tests were normal. Similarly, tests for myoglobinuria and erythrocytic dysmorphism were also negative, and the proteinuria dosage was normal.
After the first pulse of corticotherapy, the urea level was 77mg/dL and Scr was at 4.3mg/dL (ClCr 26.6mL/min/1.73m2SC). On the third day of pulse therapy, urea was at 100mg/dL, with Scr at 3.6mg/dL, ClCr at 31,8mL/min/1.73m2SC, and diuresis at 2.2ml/kg/hour. It was decided to suspend pulse therapy and start treatment with administration of prednisone at a dose of 80mg/day. After 48 hours, there was already a significant improvement in renal function, with Scr at 2.2mg/dL and urea at 86mg/dL (ClCr 52.1ml/min/1,73m²SC).
The anatomopathological study of the renal biopsy, which was performed 21 days after the start of the treatment with pulse therapy and suspension of the drug, showed slight and non-specific changes, meaning the presence of tubules with slight swelling, congested vessels, and preserved interstitium. There was no inflammatory neither fibrotic component identified in the sample.
In the outpatient control, 30 days after discharge from hospital, while using prednisone at 80mg/day, the patient was in good health and his kidney function had returned to normal; his Scr level was 1.0mg/dL and his urea was at 53mg/dl (ClCr of 114.8mL/min/1.73m2 SC, and pRIFLE of more than 100%).
DISCUSSION
The study presented clinical data of a 14-year-old boy, who had suffered multiple traumas, and who had developed AKI through ATIN, due to the use of phenytoin. Here we stress the progressive and acute nature of the worsening of his renal function (Graph 1), as this was a patient who had been in perfect previous health, without any history of glomerulopathies or auto-immune diseases. Other possibilities that were ruled out [Immediately right away] include: crush syndrome, rhabdomyolysis, compartment syndrome, shock with harm to haemodynamics, and sepsis, which could have happened because of the accident [3].
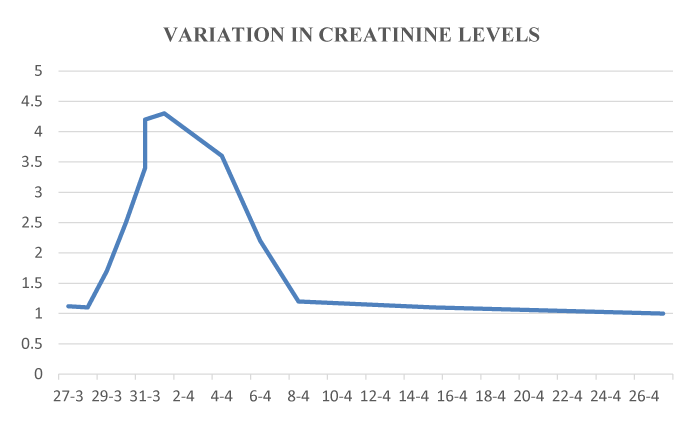
Graph 1: Variation of creatinine levels since first admittance to hospital, during the treatment, and also after discharge from hospital while under outpatient control. The peak shows the moment when phenytoin was discontinued and corticoid therapy was started.
After widespread clinical, laboratory and image appraisal of the possible pre-renal, renal, or post-renal causes of AKI were ruled out. The patient had a CT scan on admission to hospital, without radiological contrast, and throughout his hospital stay, he never received antibiotics, vasoactive drugs or NSAIDs, which are the substances most commonly connected to AKI [7-11]. It is important to note that the initial leukocytosis normalized rapidly, proving that it resulted of syndrome of the inflammatory response to stress [3]. It was considered that the AKI could have originated from medication secondary to the use of phenytoin, which, according to some previous related cases, could lead to nephrotoxicity [8,12-16. This means that the drug was immediately stopped and, due to the steady worsening of renal function, therapy based on corticosteroids was then promptly started [4,5,17,18].
Patients with ATIN resulting from medication, in about 20% of cases, can show the traditional triad of fever, eosinophilia and skin rash. However, the lack of these symptoms, as in the case reported, does not [in itself] rule out this possibility [18,5]. The picture of an ATIN-related renal failure can be completely asymptomatic or accompanied by some clinical or laboratory findings that, when identified, are very important to guide to the diagnosis [19].
Analysis of the urine can show differing degrees of proteinuria, whether associated with pyuria and haematuria or not [6,7,18]. In our case, the patient had no significant urinary alterations beyond gross haematuria hours after the introduction of the bladder catheter and, persistence of microscopic haematuria without dysmorphic erythrocytes during his hospitalization - even after removal of the bladder catheter and performed an abdominal ultrasound without abnormal findings. It is true that in some cases, bleeding from urinary tract may lead to obstructive uropathy and AKI. However, in this scenario, it would be expect hydronephrosis on ultrasound and or a hyperdense thickening of the renal pelvis and ureters, with narrowing of the lumen by the CT [20].
Recent report about the analysis of the urinary sediment in NTIA cases, surprisingly found that: RBC casts were noted in 26% of the cases and WBC casts in only 14% of the case of drug-induced ATIN [21]. Furthermore, although urine concentrations of sodium (Na), urea and creatinine either examined alone or as fractional excretions (FE) of Na (FENa) or urea (FEurea) are largely used to asses patients with AKI, save some exceptions, they just have greatest utility in distinguish pre-renal AKI from acute tubular necrosis (ATN), but they are not useful in evaluation of ATIN (Perazella 2014).
The WBC casts in the urine of our patient were normal and there was any sign of eosinophilia or eosinophiluria. Some recent studies show that, as with other tests employed in the evaluation of ATIN, serum eosinophils are not a sensitive finding. They may be only slightly elevated or markedly abnormal, at times making up 50-75% of the total WBC count. As with fever and drug rash, significant eosinophilia in ATIN has a wide range, is more common with specific drugs (similar to drug rash), and may be absent even when an eosinophil-dominant ATIN is seen on kidney biopsy (Perazella, 2014). Most disappointing is the lack of diagnostic utility of the combination of fever, rash, and eosinophilia for ATIN, where the triad is seen in no more than 20% of the cases Perazella, 2014 [5].
Microscopic, and less commonly macroscopic, haematuria is frequently observed in about 50% of the cases. Pyuria is considered a common urinary abnormality in the setting of drug-induced ATIN. In recent reports on methicillin-associated ATIN, leukocytes were noted to be practically always present. However, in other forms of drug-induced ATIN, leukocytes are noted in about 50% or even less of the cases. Although there are many studies that confirm haematuria and leukocyturia as common finding; Physicians should not mistakenly exclude ATIN as a cause of AKI in the absence of either haematuria or sterile pyuria (with or without eosinophils) [18,21].
The diagnosis of ATIN shall be considered for hospitalised patients who show a steady and unexplained rise in serum creatinine, which in most cases characterises a case of non-oliguric AKI (Perazella, 2014). In some patients, as in the present case, AKI can be serious enough to show rapid worsening of the patient’s health, together with oliguria [18]. The latent period may be as short as 1 day after some antibiotics or as long as several months with NSAIDs [19].
ATIN resulting from the use of medication is probably immunologically mediated and idiosyncratic in nature [6]. Several patients make use of drugs that could potentially lead to ATIN, but only a small percentage of these patients actually develop the disease-some of these individuals, however, also show other extra-renal signs of hypersensitivity and the effects are not dose-dependent [22]. There is no consensus about the time between exposure to the drug and the development of ATIN. There are reports that mention intervals of 1 to 5 days [23], while others mention much longer periods [7,8]. So far, in specialized literature, there are 5 reports addressing the issue of ATIN caused by use of phenytoin, but these reports do not mention a specific time correlation between the period of exposure to the drug and renal involvement [12-16]. One interesting fact is that, like in the present case, most of the patients related were young Blacks with an insidious increase in levels of serum creatinine [12,13,15].
Even though renal biopsy remains to be the gold standard for diagnosis, this is not actually a requirement in most cases, especially when there is visible and rapid clinical improvement after the identification and removal of the causative factor [8]. In humans, most drug-induced ATIN probably involves cell-mediated immunity, as renal biopsies usually do not disclose any immune deposits [22]. In other words, as reported by [23], the clinical appraisal of the case at hand, together with its evolution, allow the diagnosis of the situation and commencement of treatment when needed.
It must also be observed that, in the present case, the biopsy was only carried out 21 days after corticoid pulse therapy, which probably contributed to the non-specific character that was observed (figure 1). It is import to stress that the interstitial infiltrates characteristic of drug-induced ATIN are rapidly replaced by irreversible interstitial fibrosis and that early steroid treatment could avoid this fibrotic process by decreasing the severity of interstitial cellular infiltrates [17]. If this had been carried out prior to corticoid therapy, what was to be expected would be a pattern of interstitial inflammatory infiltrate, typically comprising monocytes, T lymphocytes and possibly eosinophils, together with oedema [5,10]. In this case, the examination, apart from ruling out the presence of any other parenchyma lesion, presented a morphological pattern which is consistent with satisfactory clinical and laboratorial progress.
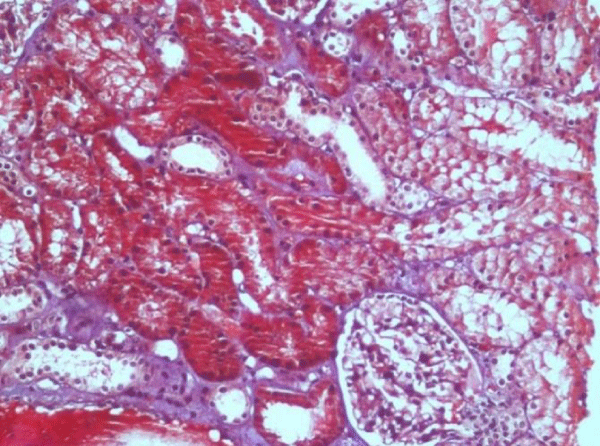
Figure 1: Discrete changes to the renal parenchyma, with general structure preserved. Glomerulus without particularities. (Masson’s trichrome stain - 200x).
CONCLUSION
ATIN is an important cause of AKI which could progress to chronic renal dysfunction [8,5]. ATIN results from injury to the tubulointerstitial tissue, which can be triggered by several different factors, such as infection, disease and illness, trauma, or several different drugs, including phenytoin [8]. It shall always be suspected in patients who show a sudden increase in levels of serum creatinine without apparent cause Perazella, 2014 [18]. The use of corticoids remains shrouded in controversy, but has been widely used in cases with important deterioration of kidney function, and has shown favourable results [4,17]. Therefore, the corticoid should be started promptly after the diagnosis of drug-induced ATIN is established to avoid the progressive replacement of interstitial cellular infiltrates by interstitial fibrosis [17]. In conclusion, the early diagnosis allows the suspension of the drug and, in the absence of a response, the application of corticosteroids, leading to normalisation of kidney function [8,23].
REFERENCES
- Freire KM, Bresolin NL, Farah AC, Carvalho FL, Góes JE. Acute kidney injury in children: incidence and prognostic factors in critical ill patients. Rev Bras Ter Intensiva. 2010; 22: 166-174. Ref.: https://goo.gl/mE7zPK
- Andreoli SP. Acute kidney injury in children. Pediatr Nephrol. 2009; 24: 253-263. Ref.: https://goo.gl/sHjdgS
- Romano TG, Tierno PF. Acute renal injury in polytrauma patients. J Bras Nefrol. 2013; 35: 48-56. Ref.: https://goo.gl/8O9mkf
- Subat-Dezulovic M, Slavić I, Rozmanić V, Persić M, Medjimurec B. Drug-induced acute tubulointerstitial nephritis: A case with elevated urinary cadmium. Pediatr Nephrol. 2002; 17: 382-285. Ref.: https://goo.gl/14eyLF
- Ulinski T, Sellier-Leclerc AL, Tudorache E, Bensman A, Aoun B. Acute tubulointerstitial nephritis. Pediatr Nephrol. 2012; 27: 1051-1057. Ref.: https://goo.gl/KH4QWI
- Rastegar A, Kashgarian M. The clinical spectrum of tubulointerstitial nephritis. Kidney Int. 1998; 59: 313-327. Ref.: https://goo.gl/o0Dh8a
- Oliveira MCD, Miguel CN. Atualização em insuficiência renal: nefrite túbulo-intersticial aguda. J Bras Nefrol. 2000; 22: 260-276.
- Kodner CM, Kudrimoti A. Diagnosis and management of acute interstitial nephritis. Am Fam Physician. 2003; 67: 2527-2534. Ref.: https://goo.gl/NqZBhe
- Tong JE, Howell DN, Foreman JW. Drug-induced granulomatous interstitial nephritis in a pediatric patient. Pediatr Nephrol. 2007; 22: 306-309. Ref.: https://goo.gl/PWGIOw
- Pannu N, Nadim MK. An overview of drug-induced acute kidney injury. Crit Care Med. 2008; 36: 216-223. Ref.: https://goo.gl/DBl5kF
- Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012; 380: 756-766. Ref.: https://goo.gl/v4Xjl3
- Sheth KJ, Casper JT, Good TA. Interstitial nephritis due to phenytoin hypersensitivity. J Pediatr. 1977; 91: 438-441. Ref.: https://goo.gl/ldVhbm
- Hyman LR, Ballow M, Knieser MR. Diphenylhydantoin interstitial nephritis. Roles of cellular and humoral immunologic injury. J Pediatr. 1978; 92: 915-920. Ref.: https://goo.gl/KcbDmS
- Hoffman EW. Phenytoin-induced interstitial nephritis. South Med J. 1981; 74: 1160-1161. Ref.: https://goo.gl/WjsnGS
- Matson JR, Krous HF, Blackstock R. Diphenylhydantoin-induced hypersensitivity reaction with interstitial nephritis. Hum Pathol. 1985; 16: 94-97. Ref.: https://goo.gl/8VoAqE
- Ram R, Swarnalatha G, Prasad N, Prayaga A, Dakshina Murthy KV. Granulomatous interstitial nephritis after prolonged use of Phenytoin. Saudi J Kidney Dis Transpl. 2009; 20: 131-133. Ref.: https://goo.gl/L4MLjy
- González E, Gutiérrez E, Galeano C, Chevia C, de Sequera P. Early steroid treatment improves the recovery of renal function in patients with drug-induced acute interstitial nephritis. Kidney Int. 2008; 73: 940-946. Ref.: https://goo.gl/inOP2n
- Perazella MA and Markowitz GS. Drug-induced acute interstitial nephritis. Nat Rev Nephrol. 2010; 6: 461-470. Ref.: https://goo.gl/V4NF1i
- Praga M and Gonzalez E. Acute interstitial nephritis. Kidney Int. 2010: 77: 956-961. Ref.: https://goo.gl/FPN5v2
- Lim AKH. Haematuria and acute kidney injury associated with warfarin anticoagulation. Gen Med (Los Angel) 2013; 1: 105. Ref.: https://goo.gl/8s2Ekq
- Fogazzi GB, Ferrari B, Garigali G, Simonini P, Consonni D. Urinary sediment findings in acute interstitial nephritis. Am J Kidney Dis 2012; 60: 330-332. Ref.: https://goo.gl/8lvGu6
- Rossert J. Drug-induced acute interstitial nephritis. Kidney Int. 2001; 60: 804-817. Ref.: https://goo.gl/mnlISR
- Patzer L. Nephrotoxicity as a cause of acute kidney injury in children. Pediatr Nephrol 2008; 23: 2159-2173. Ref.: https://goo.gl/1o0Hok